

Quantifying the contribution of sequence variants with regulatory and evolutionary significance to 34 bovine complex traits
- PMID: 31501319
- PMCID: PMC6765237
- DOI: 10.1073/pnas.1904159116
Quantifying the contribution of sequence variants with regulatory and evolutionary significance to 34 bovine complex traits
Abstract
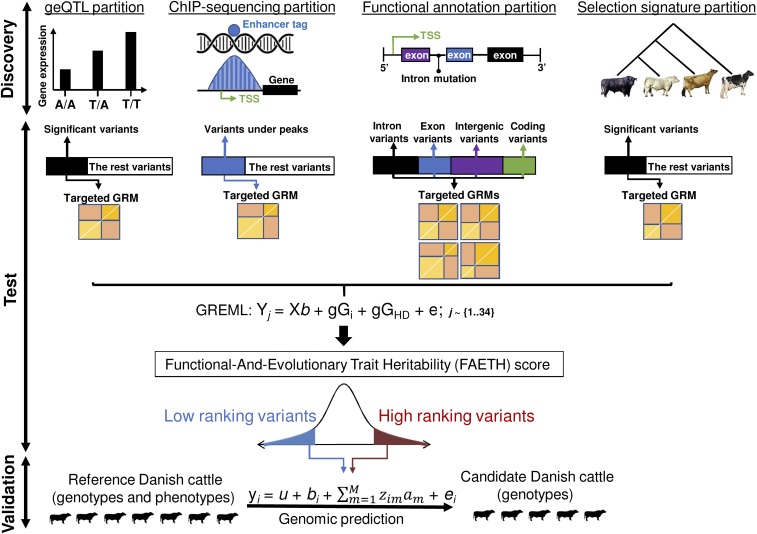
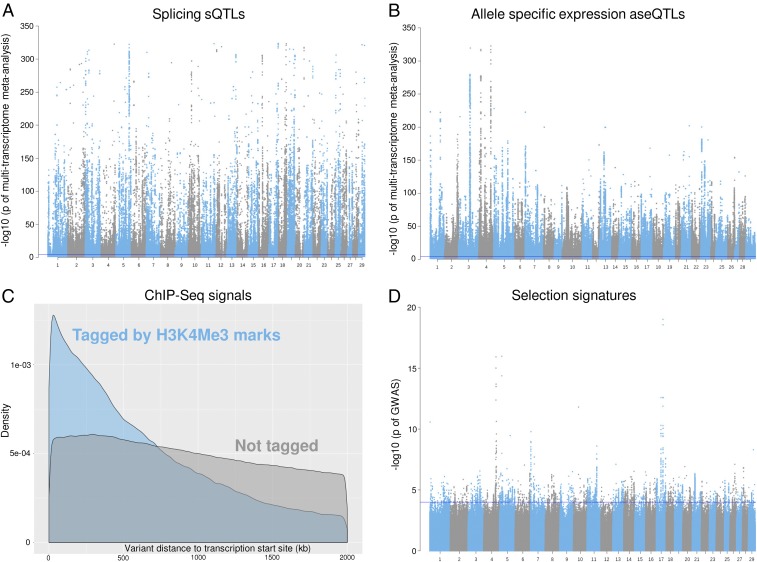
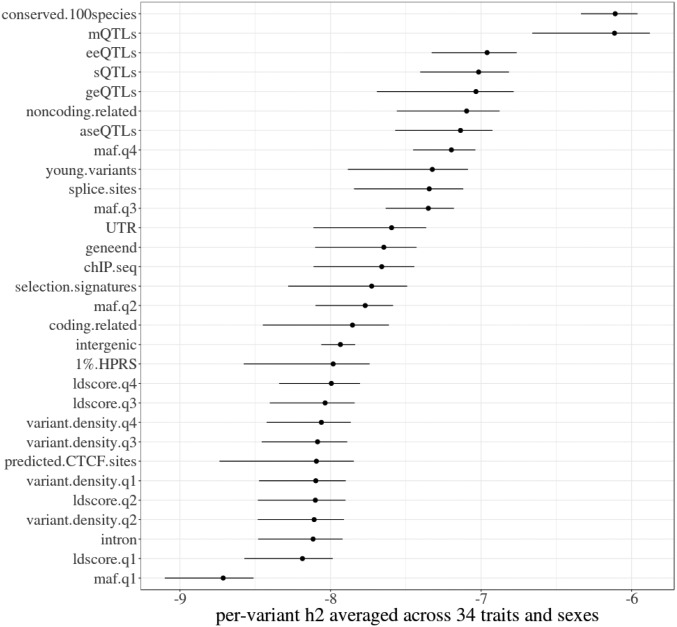
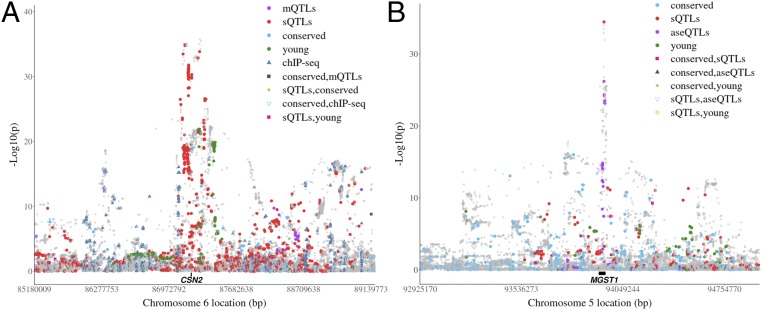
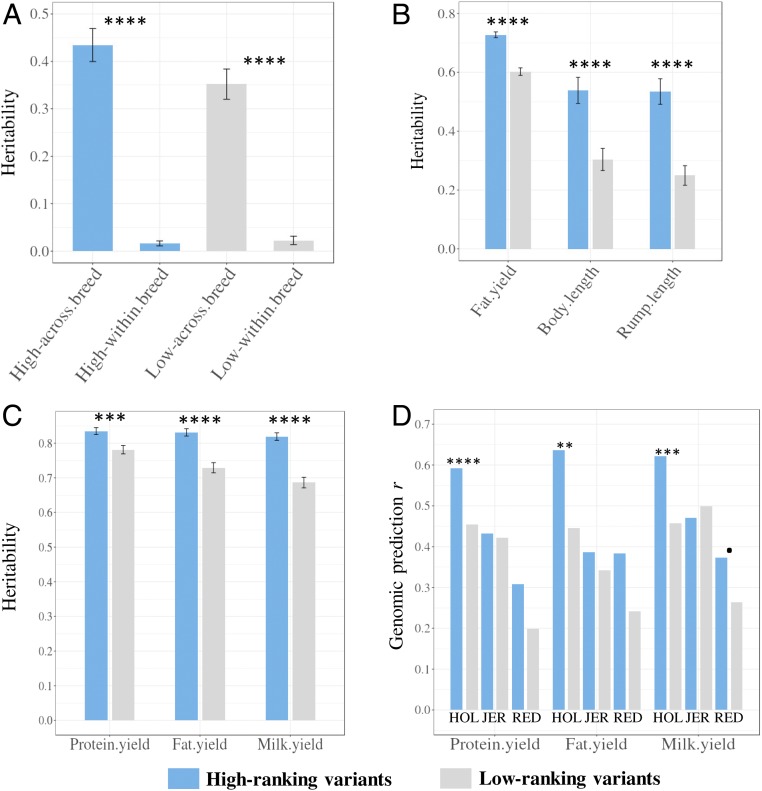
Many genome variants shaping mammalian phenotype are hypothesized to regulate gene transcription and/or to be under selection. However, most of the evidence to support this hypothesis comes from human studies. Systematic evidence for regulatory and evolutionary signals contributing to complex traits in a different mammalian model is needed. Sequence variants associated with gene expression (expression quantitative trait loci [eQTLs]) and concentration of metabolites (metabolic quantitative trait loci [mQTLs]) and under histone-modification marks in several tissues were discovered from multiomics data of over 400 cattle. Variants under selection and evolutionary constraint were identified using genome databases of multiple species. These analyses defined 30 sets of variants, and for each set, we estimated the genetic variance the set explained across 34 complex traits in 11,923 bulls and 32,347 cows with 17,669,372 imputed variants. The per-variant trait heritability of these sets across traits was highly consistent (r > 0.94) between bulls and cows. Based on the per-variant heritability, conserved sites across 100 vertebrate species and mQTLs ranked the highest, followed by eQTLs, young variants, those under histone-modification marks, and selection signatures. From these results, we defined a Functional-And-Evolutionary Trait Heritability (FAETH) score indicating the functionality and predicted heritability of each variant. In additional 7,551 cattle, the high FAETH-ranking variants had significantly increased genetic variances and genomic prediction accuracies in 3 production traits compared to the low FAETH-ranking variants. The FAETH framework combines the information of gene regulation, evolution, and trait heritability to rank variants, and the publicly available FAETH data provide a set of biological priors for cattle genomic selection worldwide.
Keywords: animal breeding; cattle; evolution; gene regulation; quantitative traits.
Copyright © 2019 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Food and Agriculture Organisation of the United Nations, FAOSTAT http://www.fao.org/faostat/en/#search/Cattle. Accessed 31 August 2018.
-
- Bouwman A. C., et al. , Meta-analysis of genome-wide association studies for cattle stature identifies common genes that regulate body size in mammals. Nat. Genet. 50, 362–367 (2018). - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
