

Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression
- PMID: 31501331
- PMCID: PMC6765259
- DOI: 10.1073/pnas.1912459116
Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression
Abstract
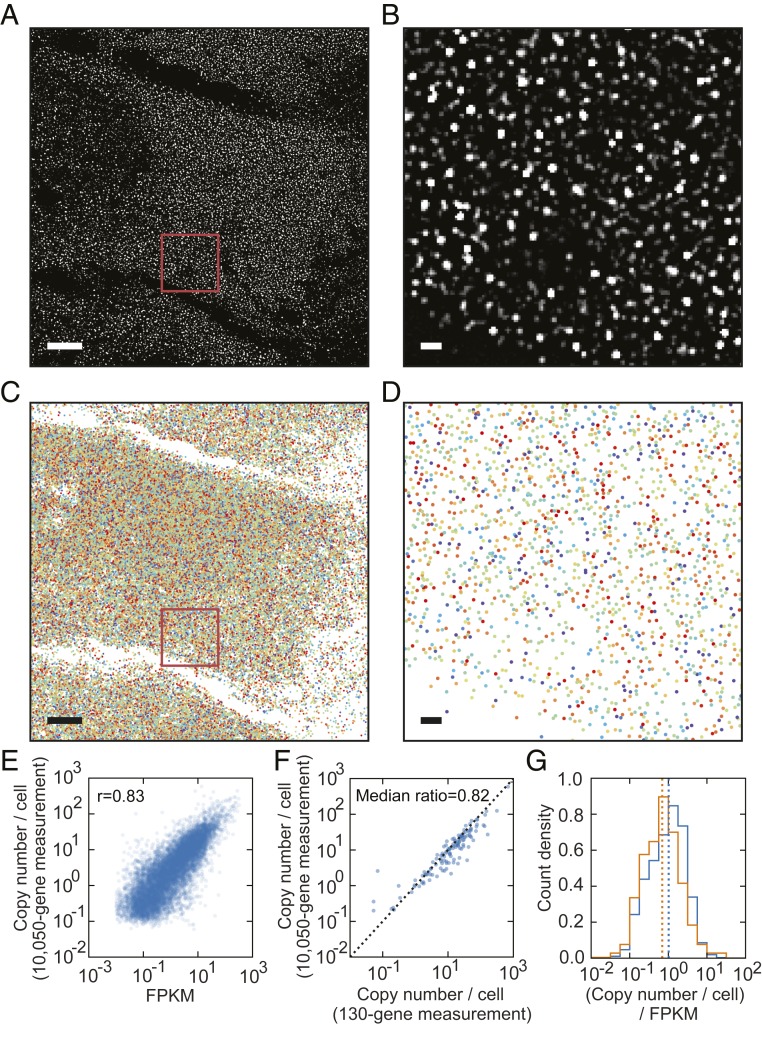
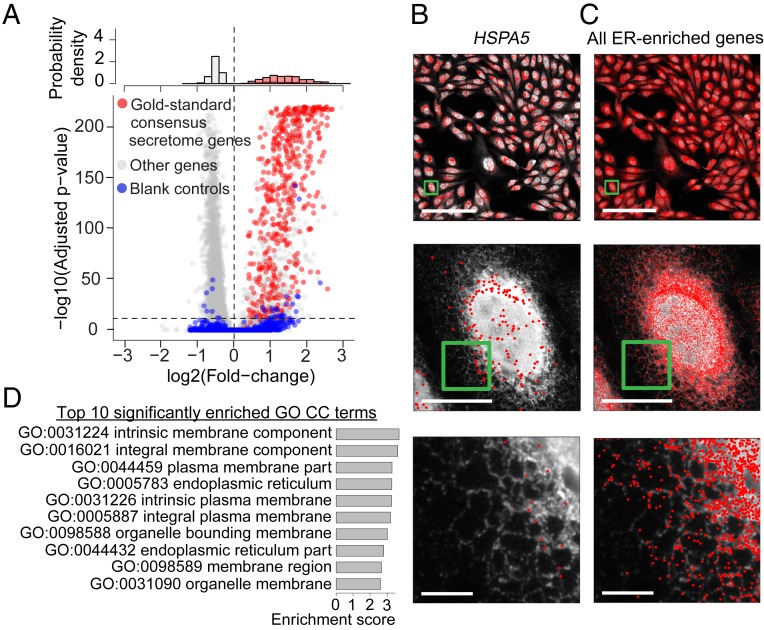
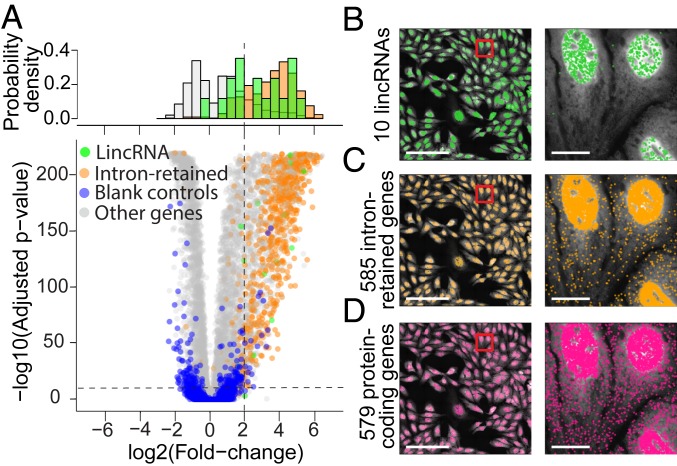
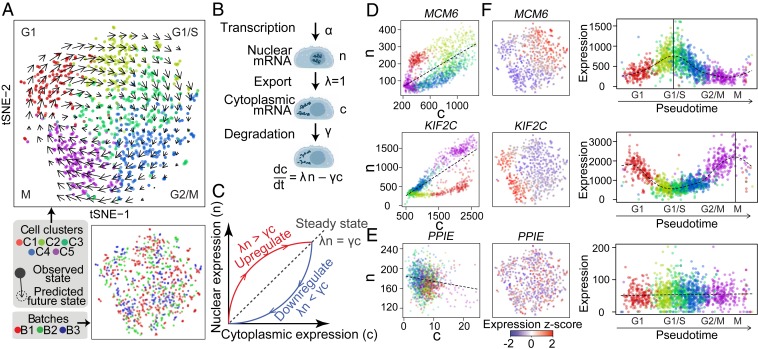
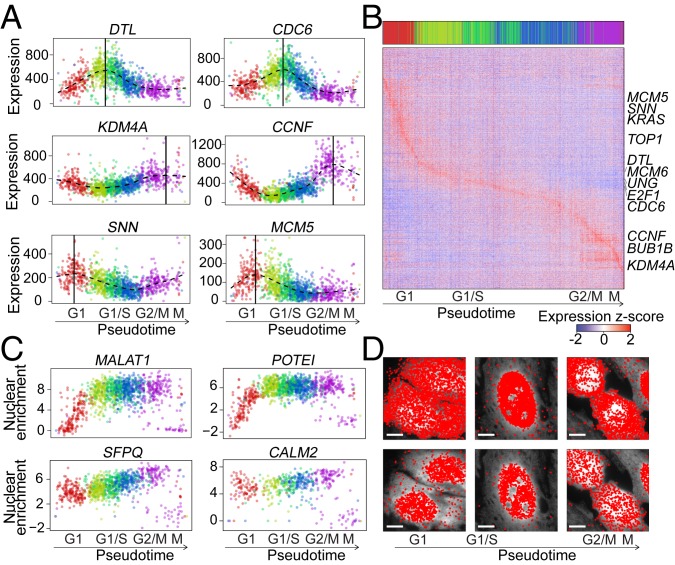
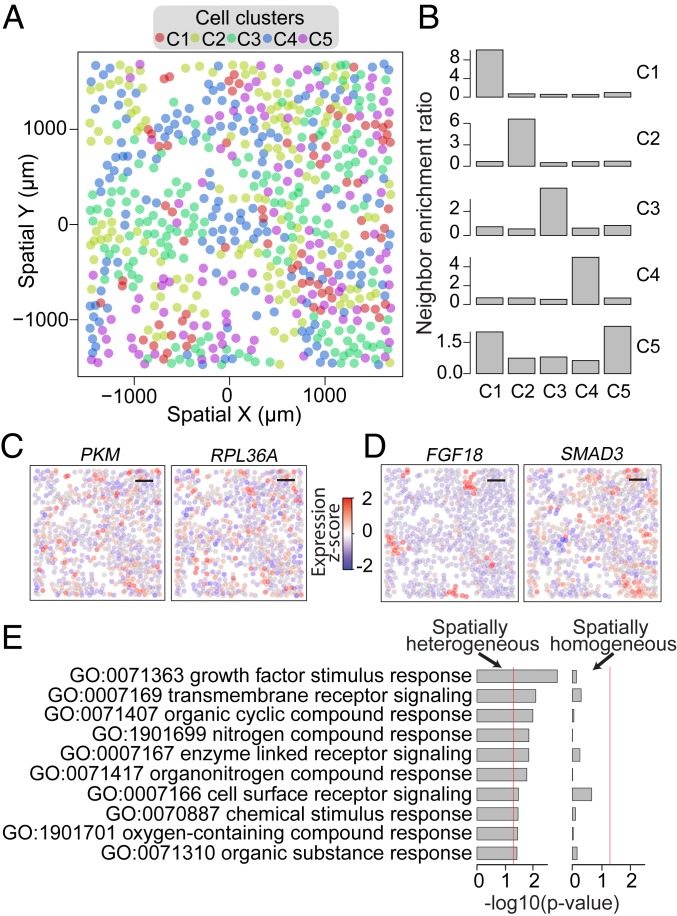
The expression profiles and spatial distributions of RNAs regulate many cellular functions. Image-based transcriptomic approaches provide powerful means to measure both expression and spatial information of RNAs in individual cells within their native environment. Among these approaches, multiplexed error-robust fluorescence in situ hybridization (MERFISH) has achieved spatially resolved RNA quantification at transcriptome scale by massively multiplexing single-molecule FISH measurements. Here, we increased the gene throughput of MERFISH and demonstrated simultaneous measurements of RNA transcripts from ∼10,000 genes in individual cells with ∼80% detection efficiency and ∼4% misidentification rate. We combined MERFISH with cellular structure imaging to determine subcellular compartmentalization of RNAs. We validated this approach by showing enrichment of secretome transcripts at the endoplasmic reticulum, and further revealed enrichment of long noncoding RNAs, RNAs with retained introns, and a subgroup of protein-coding mRNAs in the cell nucleus. Leveraging spatially resolved RNA profiling, we developed an approach to determine RNA velocity in situ using the balance of nuclear versus cytoplasmic RNA counts. We applied this approach to infer pseudotime ordering of cells and identified cells at different cell-cycle states, revealing ∼1,600 genes with putative cell cycle-dependent expression and a gradual transcription profile change as cells progress through cell-cycle stages. Our analysis further revealed cell cycle-dependent and cell cycle-independent spatial heterogeneity of transcriptionally distinct cells. We envision that the ability to perform spatially resolved, genome-wide RNA profiling with high detection efficiency and accuracy by MERFISH could help address a wide array of questions ranging from the regulation of gene expression in cells to the development of cell fate and organization in tissues.
Keywords: MERFISH; RNA velocity; fluorescence in situ hybridization; single-cell transcriptomics; spatial transcriptomics.
Copyright © 2019 the Author(s). Published by PNAS.
Conflict of interest statement
Conflict of interest statement: X.Z. is an inventor on patents applied for by Harvard University related to MERFISH.
Figures
References
-
- Engreitz J. M., Ollikainen N., Guttman M., Long non-coding RNAs: Spatial amplifiers that control nuclear structure and gene expression. Nat. Rev. Mol. Cell Biol. 17, 756–770 (2016). - PubMed
-
- Crosetto N., Bienko M., van Oudenaarden A., Spatially resolved transcriptomics and beyond. Nat. Rev. Genet. 16, 57–66 (2015). - PubMed
-
- Lein E., Borm L. E., Linnarsson S., The promise of spatial transcriptomics for neuroscience in the era of molecular cell typing. Science 358, 64–69 (2017). - PubMed
-
- Levsky J. M., Shenoy S. M., Pezo R. C., Singer R. H., Single-cell gene expression profiling. Science 297, 836–840 (2002). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
