

Biallelic mutations in neurofascin cause neurodevelopmental impairment and peripheral demyelination
- PMID: 31501903
- PMCID: PMC6763744
- DOI: 10.1093/brain/awz248
Biallelic mutations in neurofascin cause neurodevelopmental impairment and peripheral demyelination
Abstract
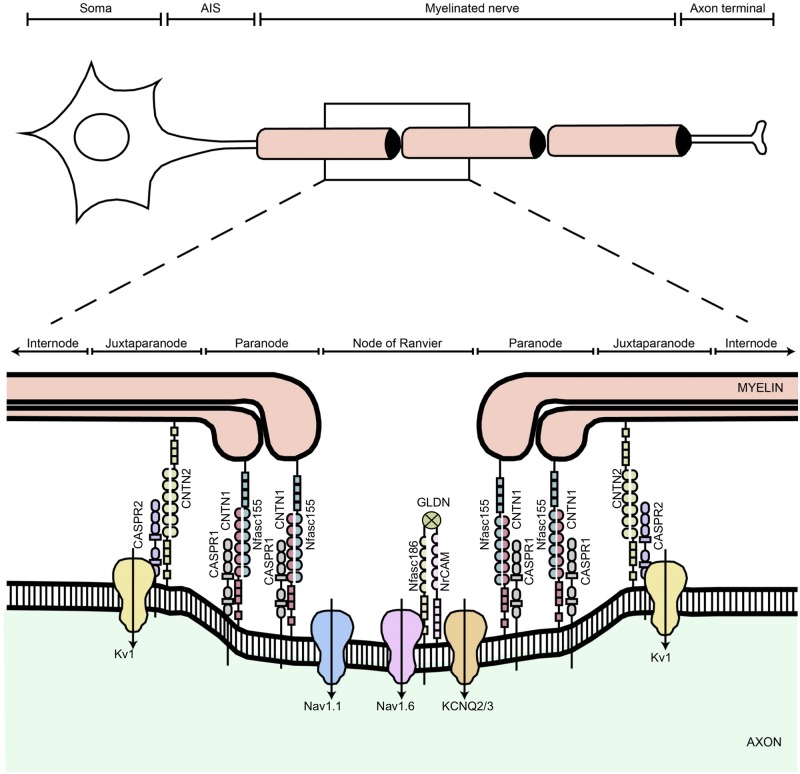
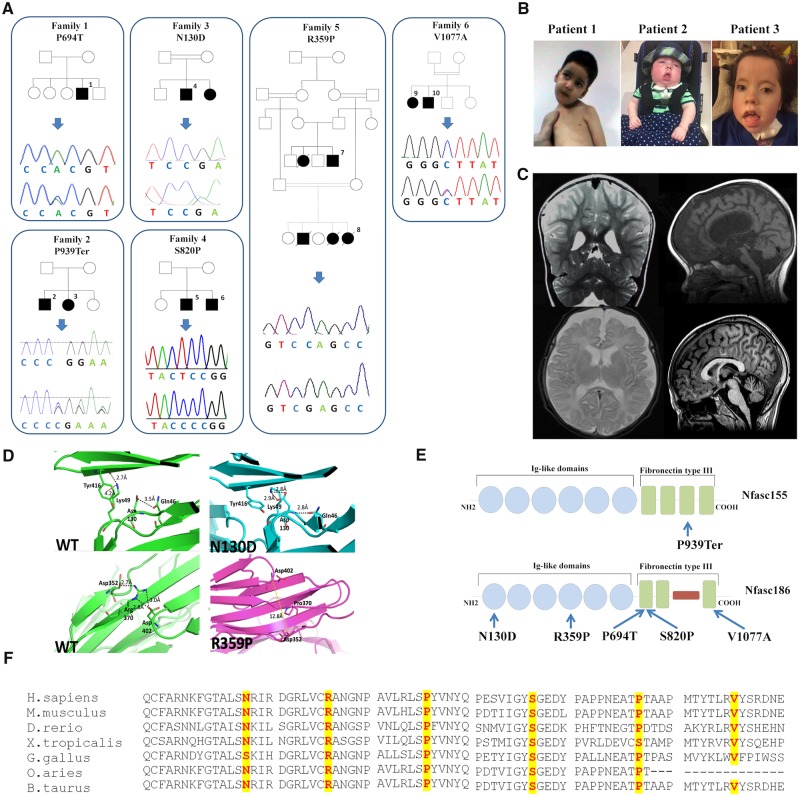
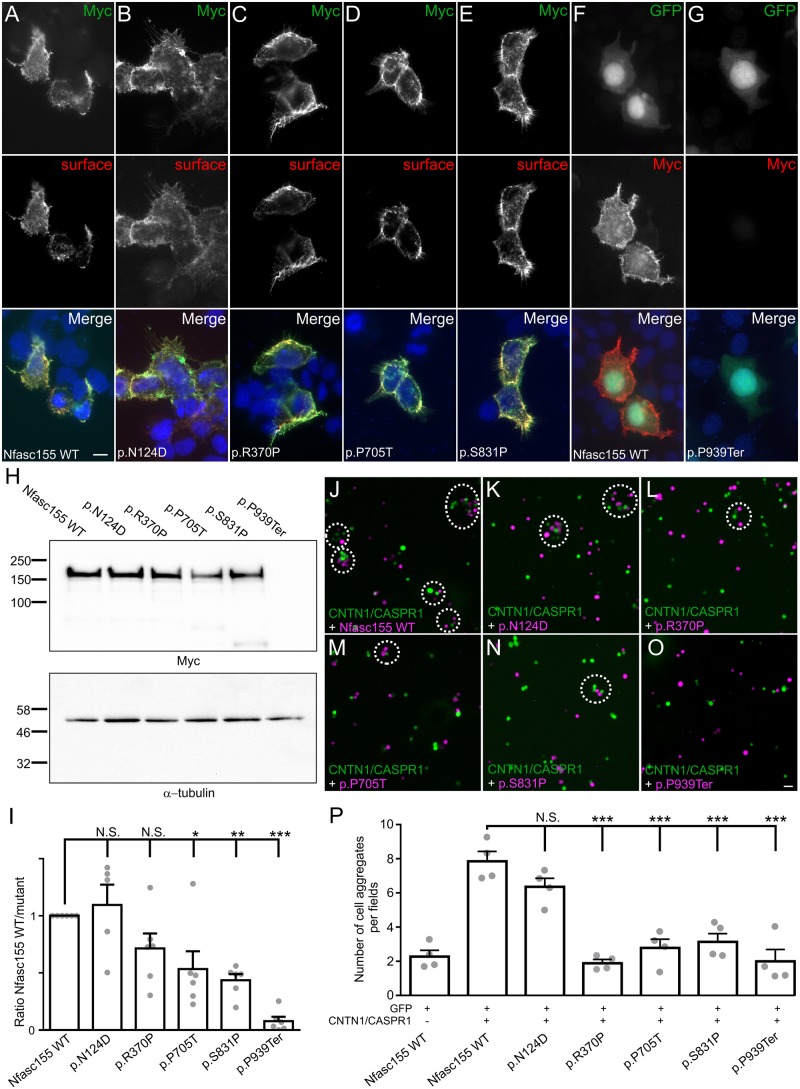
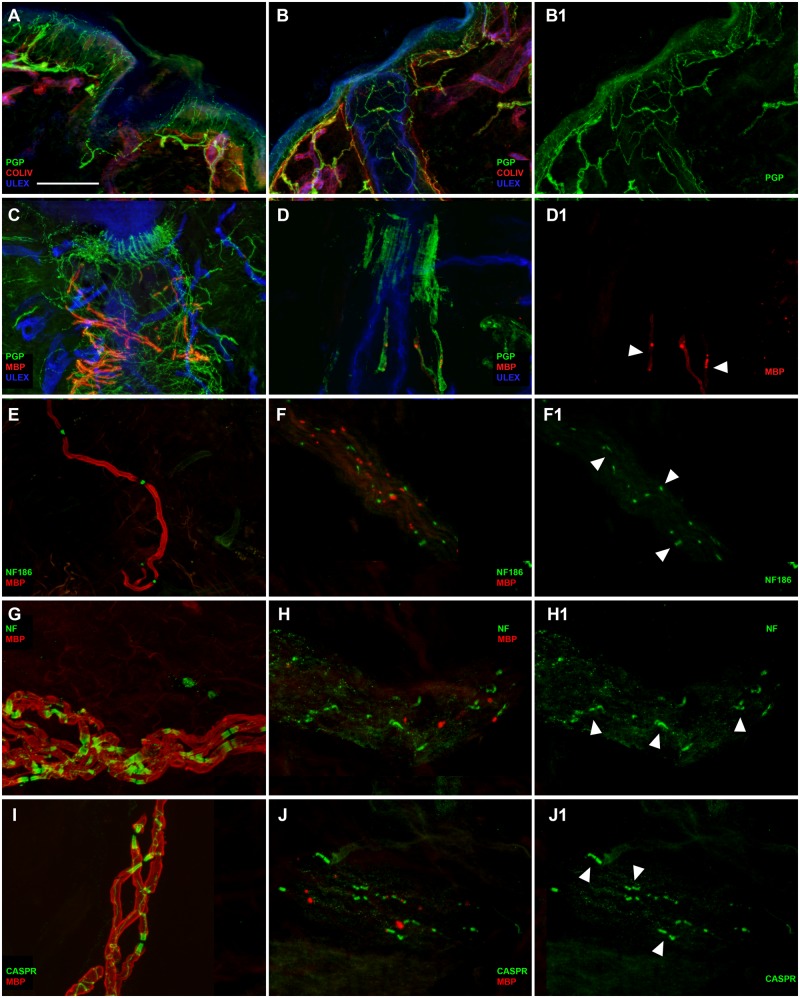
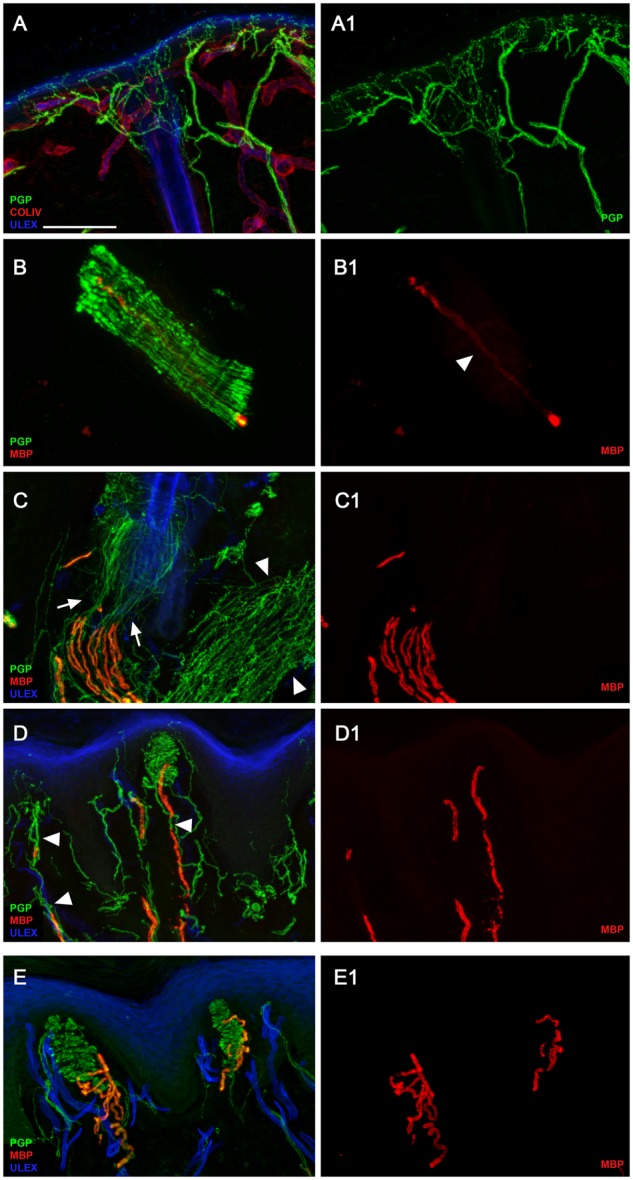
Axon pathfinding and synapse formation are essential processes for nervous system development and function. The assembly of myelinated fibres and nodes of Ranvier is mediated by a number of cell adhesion molecules of the immunoglobulin superfamily including neurofascin, encoded by the NFASC gene, and its alternative isoforms Nfasc186 and Nfasc140 (located in the axonal membrane at the node of Ranvier) and Nfasc155 (a glial component of the paranodal axoglial junction). We identified 10 individuals from six unrelated families, exhibiting a neurodevelopmental disorder characterized with a spectrum of central (intellectual disability, developmental delay, motor impairment, speech difficulties) and peripheral (early onset demyelinating neuropathy) neurological involvement, who were found by exome or genome sequencing to carry one frameshift and four different homozygous non-synonymous variants in NFASC. Expression studies using immunostaining-based techniques identified absent expression of the Nfasc155 isoform as a consequence of the frameshift variant and a significant reduction of expression was also observed in association with two non-synonymous variants affecting the fibronectin type III domain. Cell aggregation studies revealed a severely impaired Nfasc155-CNTN1/CASPR1 complex interaction as a result of the identified variants. Immunofluorescence staining of myelinated fibres from two affected individuals showed a severe loss of myelinated fibres and abnormalities in the paranodal junction morphology. Our results establish that recessive variants affecting the Nfasc155 isoform can affect the formation of paranodal axoglial junctions at the nodes of Ranvier. The genetic disease caused by biallelic NFASC variants includes neurodevelopmental impairment and a spectrum of central and peripheral demyelination as part of its core clinical phenotype. Our findings support possible overlapping molecular mechanisms of paranodal damage at peripheral nerves in both the immune-mediated and the genetic disease, but the observation of prominent central neurological involvement in NFASC biallelic variant carriers highlights the importance of this gene in human brain development and function.
Keywords: neurodevelopment; neurofascin; peripheral demyelination.
© The Author(s) (2019). Published by Oxford University Press on behalf of the Guarantors of Brain.
Figures
Comment in
-
Hereditary nodo-paranodopathies: genomic variants, not just autoantibodies, hit the protein.Brain. 2019 Oct 1;142(10):2895-2897. doi: 10.1093/brain/awz273. Brain. 2019. PMID: 31560060 No abstract available.
References
-
- Anazi S, Maddirevula S, Faqeih E, Alsedairy H, Alzahrani F, Shamseldin HE et al. . Clinical genomics expands the morbid genome of intellectual disability and offers a high diagnostic yield. Mol Psychiatry 2017; 22: 615–24. - PubMed
-
- Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys 2007; 126: 014101. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- MR/L01095X/1/MRC_/Medical Research Council/United Kingdom
- MR/K000608/1/MRC_/Medical Research Council/United Kingdom
- G1001253/MRC_/Medical Research Council/United Kingdom
- R605/0717/DMT_/The Dunhill Medical Trust/United Kingdom
- G108/638/MRC_/Medical Research Council/United Kingdom
- MR/J004758/1/MRC_/Medical Research Council/United Kingdom
- G0802760/MRC_/Medical Research Council/United Kingdom
- MR/N008324/1/MRC_/Medical Research Council/United Kingdom
- MR/S005021/1/MRC_/Medical Research Council/United Kingdom
- MR/P008399/1/MRC_/Medical Research Council/United Kingdom
- WT_/Wellcome Trust/United Kingdom
- G0601943/MRC_/Medical Research Council/United Kingdom
- MR/S01165X/1/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Miscellaneous
