

Assays to Interrogate the Ability of Compounds to Inhibit the AF-2 or AF-1 Transactivation Domains of the Androgen Receptor
- PMID: 31502857
- PMCID: PMC6921095
- DOI: 10.1089/adt.2019.940
Assays to Interrogate the Ability of Compounds to Inhibit the AF-2 or AF-1 Transactivation Domains of the Androgen Receptor
Abstract
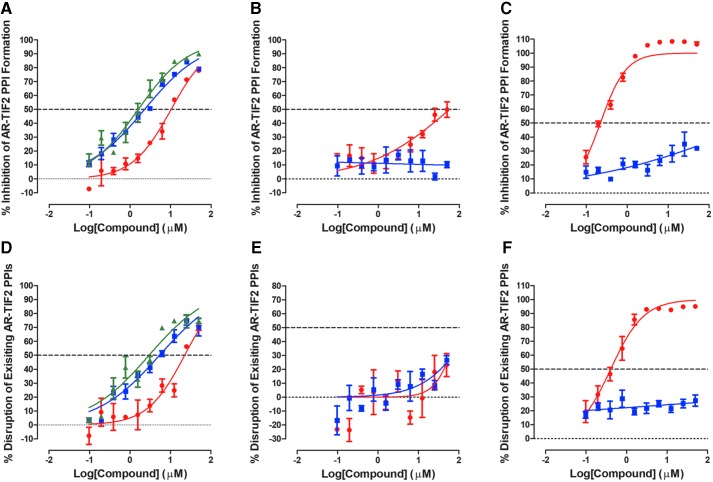
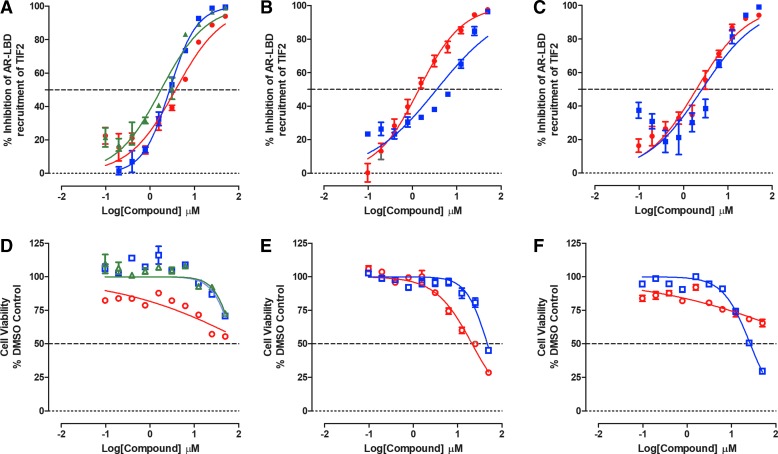
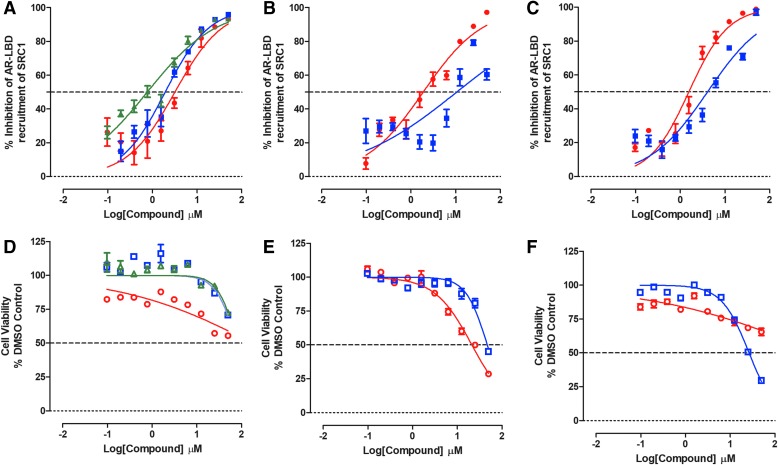
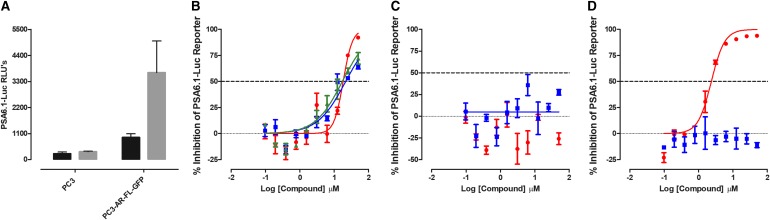
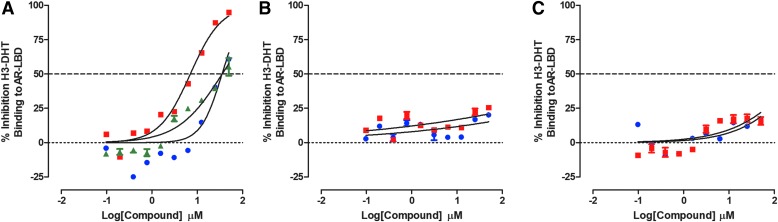
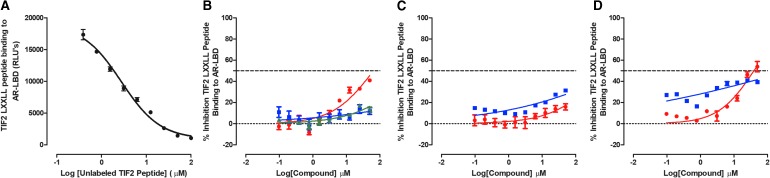
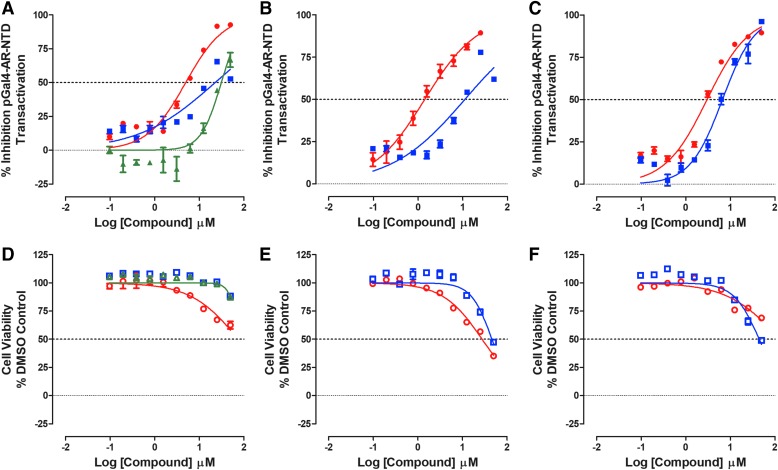
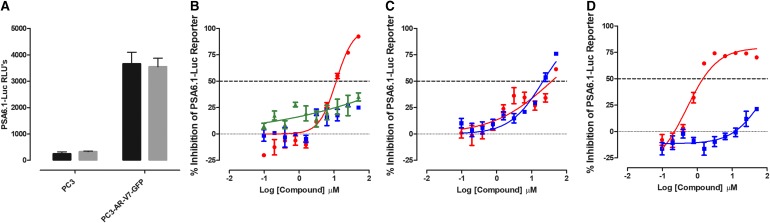
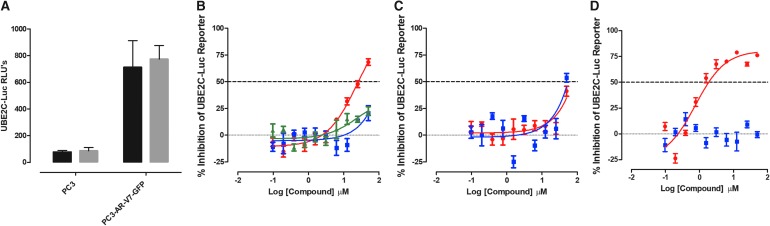
Prostate cancer is the leading cause of cancer and second leading cause of cancer-related death in men in the United States. Twenty percent of patients receiving the standard of care androgen deprivation therapy (ADT) eventually progress to metastatic and incurable castration-resistant prostate cancer (CRPC). Current FDA-approved drugs for CRPC target androgen receptor (AR) binding or androgen production, but only provide a 2- to 5-month survival benefit due to the emergence of resistance. Overexpression of AR coactivators and the emergence of AR splice variants, both promote continued transcriptional activation under androgen-depleted conditions and represent drug resistance mechanisms that contribute to CRPC progression. The AR contains two transactivation domains, activation function 2 (AF-2) and activation function 1 (AF-1), which serve as binding surfaces for coactivators involved in the transcriptional activation of AR target genes. Full-length AR contains both AF-2 and AF-1 surfaces, whereas AR splice variants only have an AF-1 surface. We have recently prosecuted a high-content screening campaign to identify hit compounds that can inhibit or disrupt the protein-protein interactions (PPIs) between AR and transcriptional intermediary factor 2 (TIF2), one of the coactivators implicated in CRPC disease progression. Since an ideal inhibitor/disruptor of AR-coactivator PPIs would target both the AF-2 and AF-1 surfaces, we describe here the development and validation of five AF-2- and three AF-1-focused assays to interrogate and prioritize hits that disrupt both transactivation surfaces. The assays were validated using a test set of seven known AR modulator compounds, including three AR antagonists and one androgen synthesis inhibitor that are FDA-approved ADTs, two investigational molecules that target the N-terminal domain of AR, and an inhibitor of the Hsp90 (heat shock protein) molecular chaperone.
Keywords: androgen receptor activation function domains; castration-resistant prostate cancer; mammalian 2-hybrid assays.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
