

From Escherichia coli mutant 13C labeling data to a core kinetic model: A kinetic model parameterization pipeline
- PMID: 31504032
- PMCID: PMC6759195
- DOI: 10.1371/journal.pcbi.1007319
From Escherichia coli mutant 13C labeling data to a core kinetic model: A kinetic model parameterization pipeline
Abstract
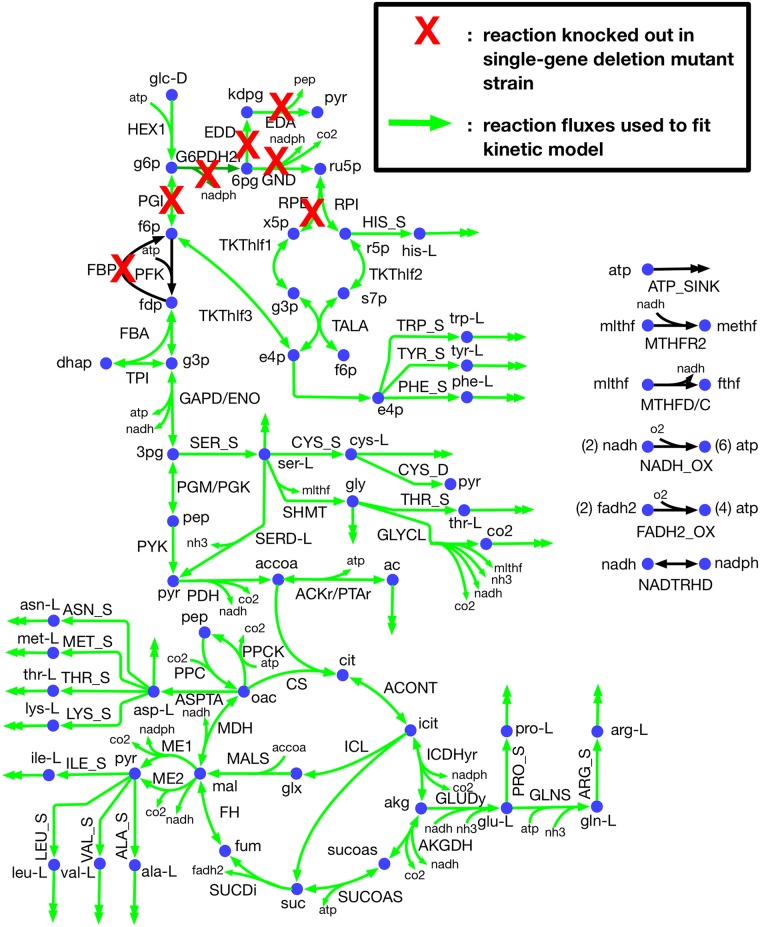
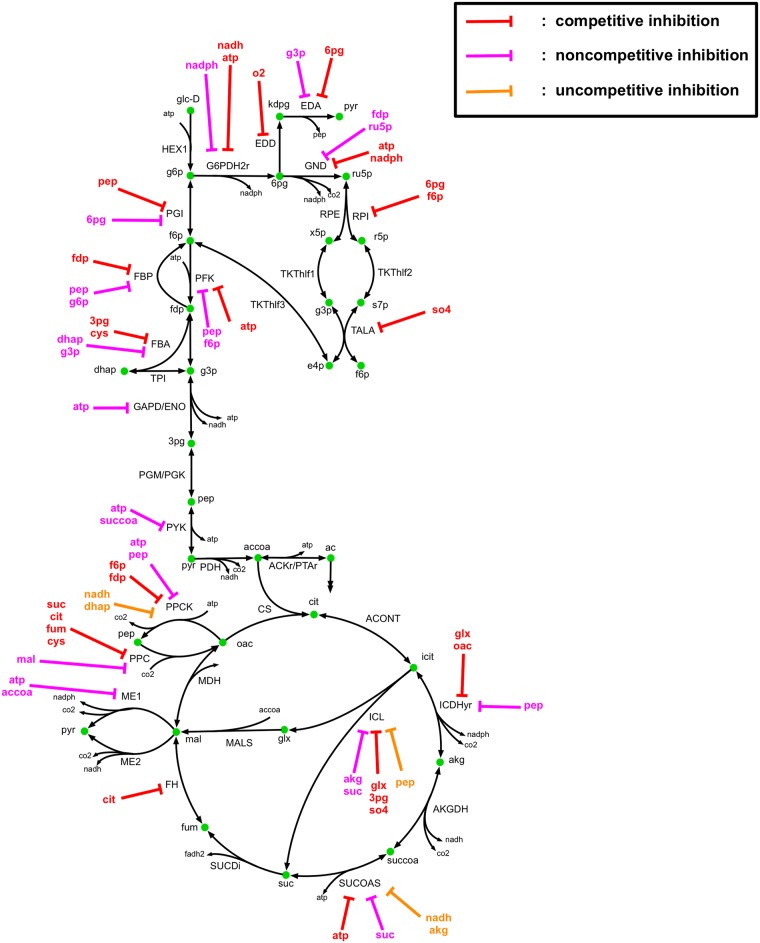
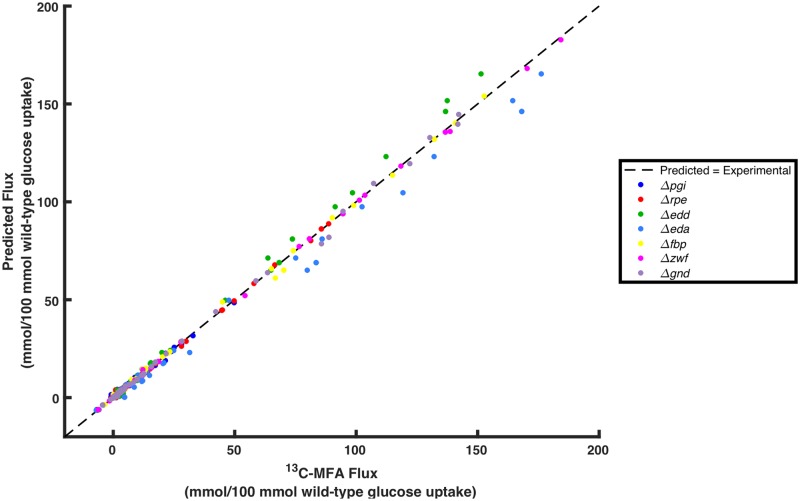
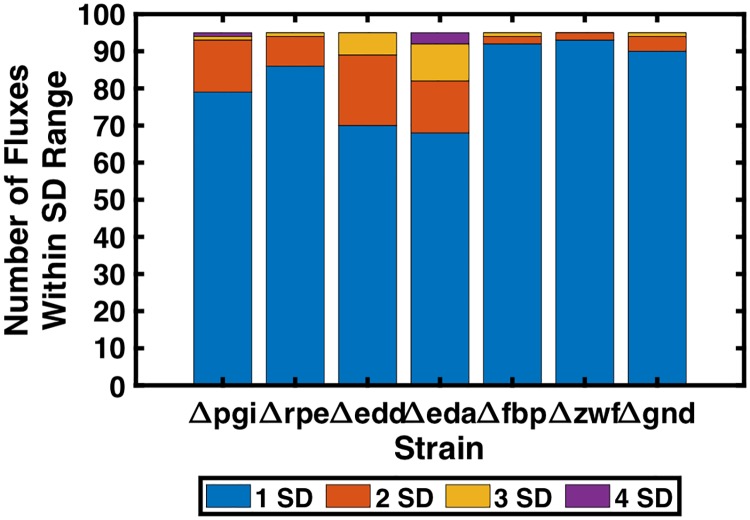
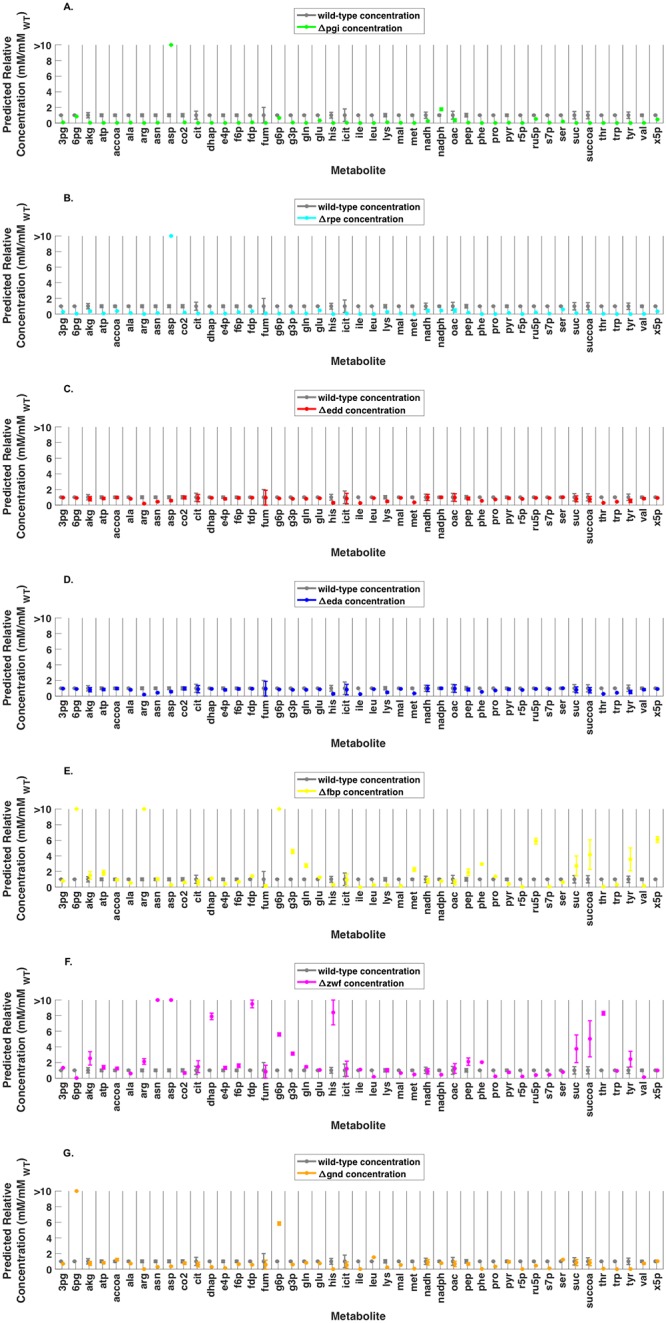
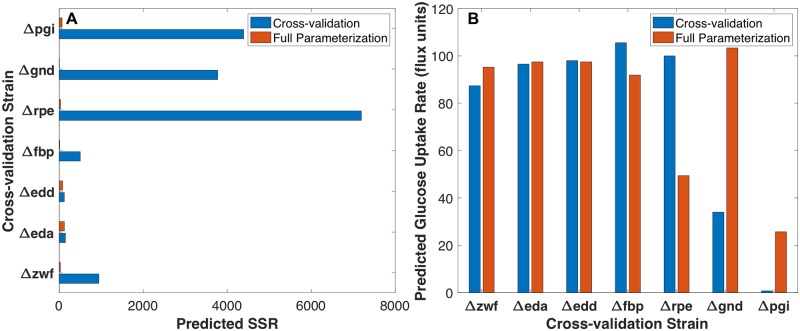
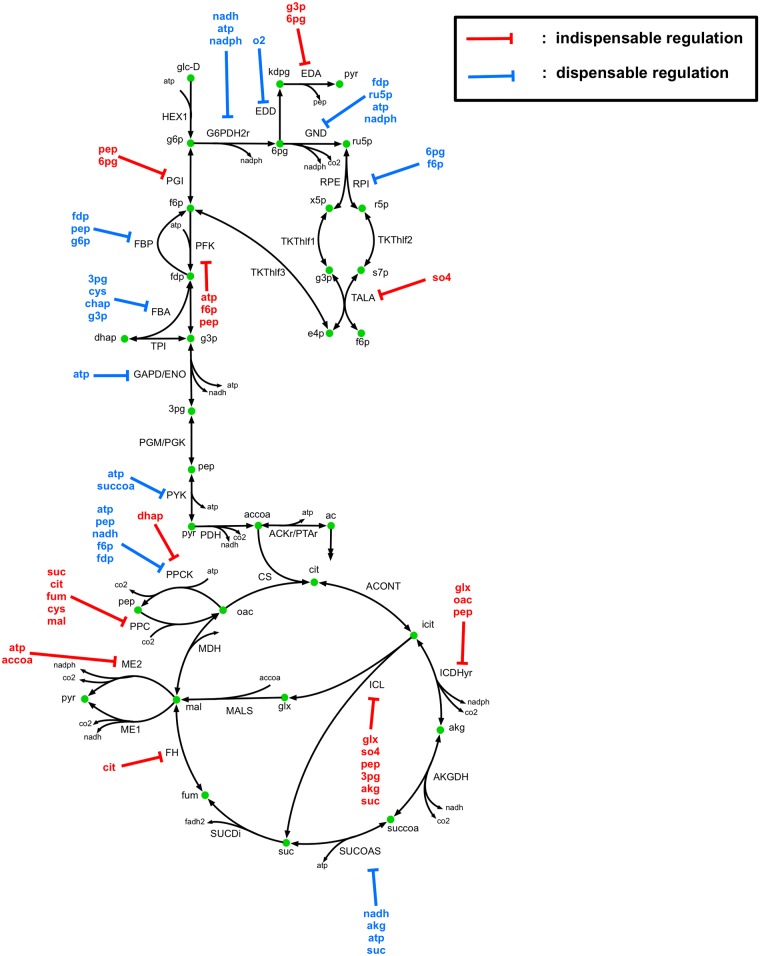
Kinetic models of metabolic networks offer the promise of quantitative phenotype prediction. The mechanistic characterization of enzyme catalyzed reactions allows for tracing the effect of perturbations in metabolite concentrations and reaction fluxes in response to genetic and environmental perturbation that are beyond the scope of stoichiometric models. In this study, we develop a two-step computational pipeline for the rapid parameterization of kinetic models of metabolic networks using a curated metabolic model and available 13C-labeling distributions under multiple genetic and environmental perturbations. The first step involves the elucidation of all intracellular fluxes in a core model of E. coli containing 74 reactions and 61 metabolites using 13C-Metabolic Flux Analysis (13C-MFA). Here, fluxes corresponding to the mid-exponential growth phase are elucidated for seven single gene deletion mutants from upper glycolysis, pentose phosphate pathway and the Entner-Doudoroff pathway. The computed flux ranges are then used to parameterize the same (i.e., k-ecoli74) core kinetic model for E. coli with 55 substrate-level regulations using the newly developed K-FIT parameterization algorithm. The K-FIT algorithm employs a combination of equation decomposition and iterative solution techniques to evaluate steady-state fluxes in response to genetic perturbations. k-ecoli74 predicted 86% of flux values for strains used during fitting within a single standard deviation of 13C-MFA estimated values. By performing both tasks using the same network, errors associated with lack of congruity between the two networks are avoided, allowing for seamless integration of data with model building. Product yield predictions and comparison with previously developed kinetic models indicate shifts in flux ranges and the presence or absence of mutant strains delivering flux towards pathways of interest from training data significantly impact predictive capabilities. Using this workflow, the impact of completeness of fluxomic datasets and the importance of specific genetic perturbations on uncertainties in kinetic parameter estimation are evaluated.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures



Similar articles
-
A kinetic model of Escherichia coli core metabolism satisfying multiple sets of mutant flux data.Metab Eng. 2014 Sep;25:50-62. doi: 10.1016/j.ymben.2014.05.014. Epub 2014 Jun 10. Metab Eng. 2014. PMID: 24928774
-
K-FIT: An accelerated kinetic parameterization algorithm using steady-state fluxomic data.Metab Eng. 2020 Sep;61:197-205. doi: 10.1016/j.ymben.2020.03.001. Epub 2020 Mar 13. Metab Eng. 2020. PMID: 32173504
-
Investigating the effects of perturbations to pgi and eno gene expression on central carbon metabolism in Escherichia coli using (13)C metabolic flux analysis.Microb Cell Fact. 2012 Jun 21;11:87. doi: 10.1186/1475-2859-11-87. Microb Cell Fact. 2012. PMID: 22721472 Free PMC article.
-
Metabolic flux analysis based on 13C-labeling experiments and integration of the information with gene and protein expression patterns.Adv Biochem Eng Biotechnol. 2004;91:1-49. doi: 10.1007/b94204. Adv Biochem Eng Biotechnol. 2004. PMID: 15453191 Review.
-
Quantitative metabolic fluxes regulated by trans-omic networks.Biochem J. 2022 Mar 31;479(6):787-804. doi: 10.1042/BCJ20210596. Biochem J. 2022. PMID: 35356967 Free PMC article. Review.
Cited by
-
Modular dynamic biomolecular modelling with bond graphs: the unification of stoichiometry, thermodynamics, kinetics and data.J R Soc Interface. 2021 Aug;18(181):20210478. doi: 10.1098/rsif.2021.0478. Epub 2021 Aug 25. J R Soc Interface. 2021. PMID: 34428949 Free PMC article.
-
Quantitative modeling of pentose phosphate pathway response to oxidative stress reveals a cooperative regulatory strategy.iScience. 2022 Jun 28;25(8):104681. doi: 10.1016/j.isci.2022.104681. eCollection 2022 Aug 19. iScience. 2022. PMID: 35856027 Free PMC article.
-
Bottom-up parameterization of enzyme rate constants: Reconciling inconsistent data.Metab Eng Commun. 2024 Apr 23;18:e00234. doi: 10.1016/j.mec.2024.e00234. eCollection 2024 Jun. Metab Eng Commun. 2024. PMID: 38711578 Free PMC article.
-
Metabolic flux analysis: a comprehensive review on sample preparation, analytical techniques, data analysis, computational modelling, and main application areas.RSC Adv. 2022 Sep 7;12(39):25528-25548. doi: 10.1039/d2ra03326g. eCollection 2022 Sep 5. RSC Adv. 2022. PMID: 36199351 Free PMC article. Review.
-
Iterative design of training data to control intricate enzymatic reaction networks.Nat Commun. 2024 Feb 21;15(1):1602. doi: 10.1038/s41467-024-45886-9. Nat Commun. 2024. PMID: 38383500 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
