

Animal Models of Normal and Disturbed Iron and Copper Metabolism
- PMID: 31504675
- PMCID: PMC6887953
- DOI: 10.1093/jn/nxz172
Animal Models of Normal and Disturbed Iron and Copper Metabolism
Abstract
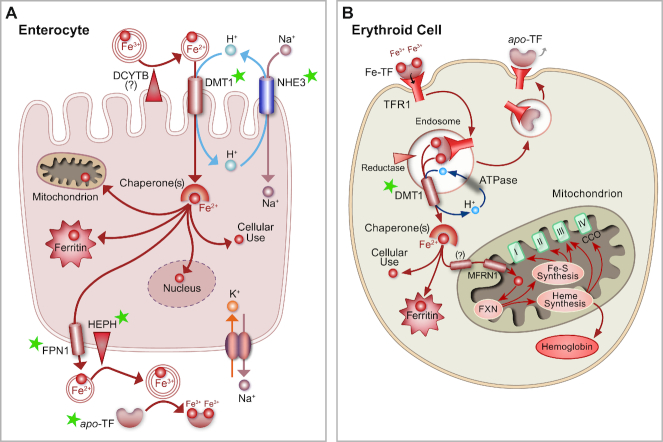
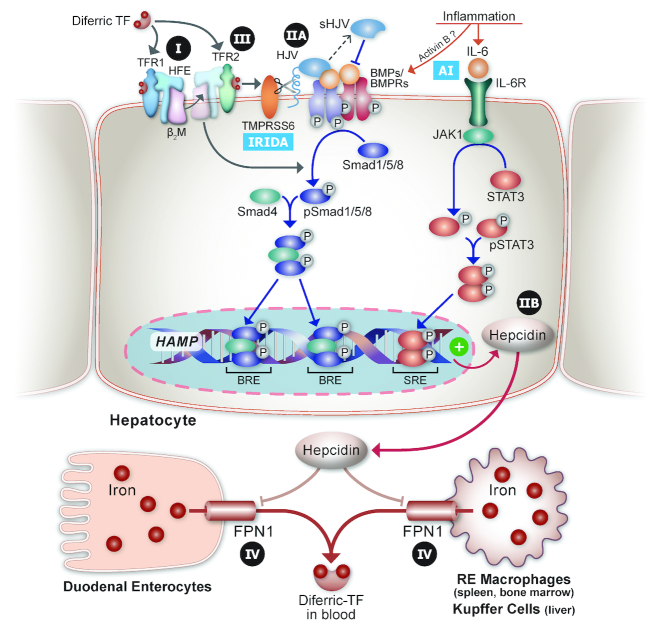
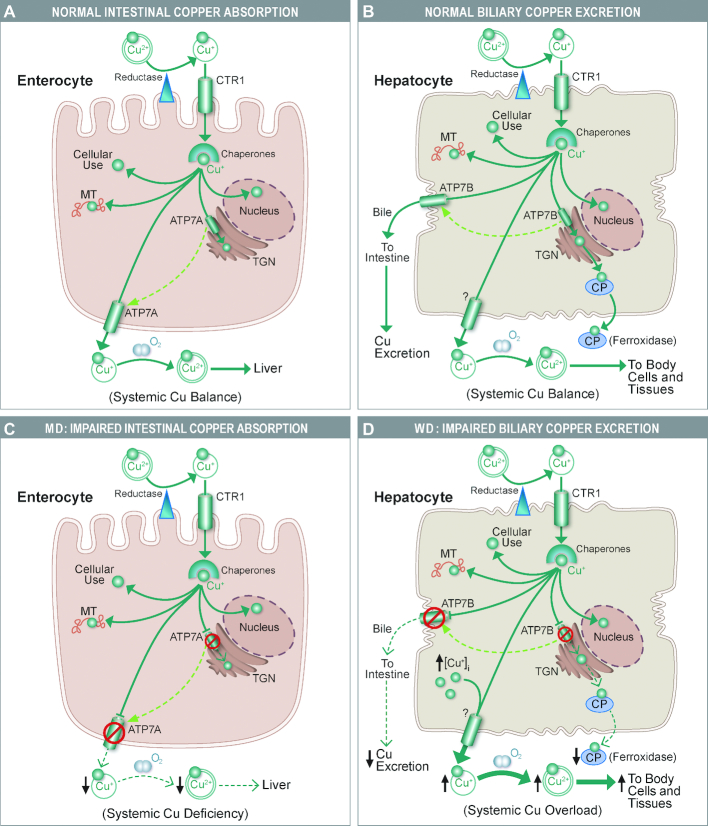
Research on the interplay between iron and copper metabolism in humans began to flourish in the mid-20th century, and diseases associated with dysregulated homeostasis of these essential trace minerals are common even today. Iron deficiency is the most frequent cause of anemia worldwide, leading to significant morbidity, particularly in developing countries. Iron overload is also quite common, usually being the result of genetic mutations which lead to inappropriate expression of the iron-regulatory hormone hepcidin. Perturbations of copper homeostasis in humans have also been described, including rare genetic conditions which lead to severe copper deficiency (Menkes disease) or copper overload (Wilson disease). Historically, the common laboratory rat (Rattus norvegicus) was the most frequently utilized species to model human physiology and pathophysiology. Recently, however, the development of genetic-engineering technology combined with the worldwide availability of numerous genetically homogenous (i.e., inbred) mouse strains shifted most research on iron and copper metabolism to laboratory mice. This created new opportunities to understand the function of individual genes in the context of a living animal, but thoughtful consideration of whether mice are the most appropriate models of human pathophysiology was not necessarily involved. Given this background, this review is intended to provide a guide for future research on iron- and copper-related disorders in humans. Generation of complementary experimental models in rats, swine, and other mammals is now facile given the advent of newer genetic technologies, thus providing the opportunity to accelerate the identification of pathogenic mechanisms and expedite the development of new treatments to mitigate these important human disorders.
Keywords: Menkes disease; Wilson disease; anemia; copper deficiency; hepcidin; hereditary hemochromatosis; iron deficiency; iron-deficiency anemia; β-thalassemia.
Copyright © American Society for Nutrition 2019.
Figures
Similar articles
-
A competitive enzyme-linked immunosorbent assay specific for murine hepcidin-1: correlation with hepatic mRNA expression in established and novel models of dysregulated iron homeostasis.Haematologica. 2015 Feb;100(2):167-77. doi: 10.3324/haematol.2014.116723. Epub 2014 Nov 25. Haematologica. 2015. PMID: 25425686 Free PMC article.
-
Iron-deficiency anemia secondary to mutations in genes controlling hepcidin.Expert Rev Hematol. 2010 Apr;3(2):205-16. doi: 10.1586/ehm.10.2. Expert Rev Hematol. 2010. PMID: 21083463 Review.
-
Mild copper deficiency alters gene expression of proteins involved in iron metabolism.Blood Cells Mol Dis. 2006 Jan-Feb;36(1):15-20. doi: 10.1016/j.bcmd.2005.11.003. Epub 2006 Jan 5. Blood Cells Mol Dis. 2006. PMID: 16406711
-
Functional iron deficiency in toxic milk mutant mice (tx-J) despite high hepatic ferroportin: a critical role of decreased GPI-ceruloplasmin expression in liver macrophages.Metallomics. 2019 Jun 19;11(6):1079-1092. doi: 10.1039/c9mt00035f. Metallomics. 2019. PMID: 31011744
-
Role of iron metabolism in heart failure: From iron deficiency to iron overload.Biochim Biophys Acta Mol Basis Dis. 2019 Jul 1;1865(7):1925-1937. doi: 10.1016/j.bbadis.2018.08.030. Epub 2018 Aug 26. Biochim Biophys Acta Mol Basis Dis. 2019. PMID: 31109456 Review.
Cited by
-
Wilson's Disease-Crossroads of Genetics, Inflammation and Immunity/Autoimmunity: Clinical and Molecular Issues.Int J Mol Sci. 2024 Aug 20;25(16):9034. doi: 10.3390/ijms25169034. Int J Mol Sci. 2024. PMID: 39201720 Free PMC article. Review.
-
Development of rat and mouse models of heme-iron absorption.JCI Insight. 2025 Jun 9;10(11):e184742. doi: 10.1172/jci.insight.184742. eCollection 2025 Jun 9. JCI Insight. 2025. PMID: 40485584 Free PMC article.
-
Putative Animal Models of Restless Legs Syndrome: A Systematic Review and Evaluation of Their Face and Construct Validity.Neurotherapeutics. 2023 Jan;20(1):154-178. doi: 10.1007/s13311-022-01334-4. Epub 2022 Dec 19. Neurotherapeutics. 2023. PMID: 36536233 Free PMC article.
-
Iron and Cadmium Entry Into Renal Mitochondria: Physiological and Toxicological Implications.Front Cell Dev Biol. 2020 Sep 2;8:848. doi: 10.3389/fcell.2020.00848. eCollection 2020. Front Cell Dev Biol. 2020. PMID: 32984336 Free PMC article. Review.
-
Iron-dependent apoptosis causes embryotoxicity in inflamed and obese pregnancy.Nat Commun. 2021 Jun 29;12(1):4026. doi: 10.1038/s41467-021-24333-z. Nat Commun. 2021. PMID: 34188052 Free PMC article.
References
-
- Zhang F, Wen Y, Guo X. CRISPR/Cas9 for genome editing: progress, implications and challenges. Hum Mol Genet. 2014;23:R40–6. - PubMed
-
- Doudna J, Mali P. CRISPR-Cas: a laboratory manual. New York: Cold Spring Harbor Laboratory Press; 2016.
-
- Homberg JR, Wohr M, Alenina N. Comeback of the rat in biomedical research. ACS Chem Neurosci. 2017;8:900–3. - PubMed
