

Monoacylglycerol Lipase Inhibition Protects From Liver Injury in Mouse Models of Sclerosing Cholangitis
- PMID: 31505038
- PMCID: PMC7317927
- DOI: 10.1002/hep.30929
Monoacylglycerol Lipase Inhibition Protects From Liver Injury in Mouse Models of Sclerosing Cholangitis
Abstract
Background and aims: Monoacylglycerol lipase (MGL) is the last enzymatic step in triglyceride degradation, hydrolyzing monoglycerides into glycerol and fatty acids (FAs) and converting 2-arachidonoylglycerol into arachidonic acid, thus providing ligands for nuclear receptors as key regulators of hepatic bile acid (BA)/lipid metabolism and inflammation. We aimed to explore the role of MGL in the development of cholestatic liver and bile duct injury in mouse models of sclerosing cholangitis, a disease so far lacking effective pharmacological therapy.
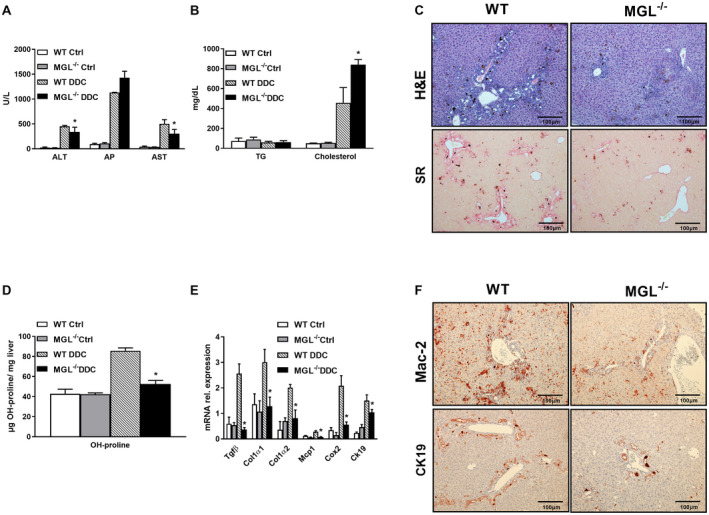
Approach and results: To this aim we analyzed the effects of 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) feeding to induce sclerosing cholangitis in wild-type (WT) and knockout (MGL-/- ) mice and tested pharmacological inhibition with JZL184 in the multidrug resistance protein 2 knockout (Mdr2-/- ) mouse model of sclerosing cholangitis. Cholestatic liver injury and fibrosis were assessed by serum biochemistry, liver histology, gene expression, and western blot characterization of BA and FA synthesis/transport. Moreover, intestinal FAs and fecal microbiome were analyzed. Transfection and silencing were performed in Caco2 cells. MGL-/- mice were protected from DDC-induced biliary fibrosis and inflammation with reduced serum liver enzymes and increased FA/BA metabolism and β-oxidation. Notably, pharmacological (JZL184) inhibition of MGL ameliorated cholestatic injury in DDC-fed WT mice and protected Mdr2-/- mice from spontaneous liver injury, with improved liver enzymes, inflammation, and biliary fibrosis. In vitro experiments confirmed that silencing of MGL decreases prostaglandin E2 accumulation in the intestine and up-regulates peroxisome proliferator-activated receptors alpha and gamma activity, thus reducing inflammation.
Conclusions: Collectively, our study unravels MGL as a metabolic target, demonstrating that MGL inhibition may be considered as potential therapy for sclerosing cholangitis.
© 2019 The Authors. Hepatology published by Wiley Periodicals, Inc., on behalf of American Association for the Study of Liver Diseases.
Figures







Similar articles
-
Inhibition of intestinal bile acid absorption improves cholestatic liver and bile duct injury in a mouse model of sclerosing cholangitis.J Hepatol. 2016 Mar;64(3):674-81. doi: 10.1016/j.jhep.2015.10.024. Epub 2015 Oct 31. J Hepatol. 2016. PMID: 26529078
-
Tetrahydroxylated bile acids improve cholestatic liver and bile duct injury in the Mdr2-/- mouse model of sclerosing cholangitis via immunomodulatory effects.Hepatol Commun. 2022 Sep;6(9):2368-2378. doi: 10.1002/hep4.1998. Epub 2022 Jun 12. Hepatol Commun. 2022. PMID: 35691019 Free PMC article.
-
Colesevelam attenuates cholestatic liver and bile duct injury in Mdr2-/- mice by modulating composition, signalling and excretion of faecal bile acids.Gut. 2018 Sep;67(9):1683-1691. doi: 10.1136/gutjnl-2017-314553. Epub 2018 Apr 10. Gut. 2018. PMID: 29636383 Free PMC article.
-
PPAR-Mediated Bile Acid Glucuronidation: Therapeutic Targets for the Treatment of Cholestatic Liver Diseases.Cells. 2024 Aug 1;13(15):1296. doi: 10.3390/cells13151296. Cells. 2024. PMID: 39120326 Free PMC article. Review.
-
Novel and emerging therapies for cholestatic liver diseases.Liver Int. 2018 Sep;38(9):1520-1535. doi: 10.1111/liv.13880. Epub 2018 Jun 14. Liver Int. 2018. PMID: 29758112 Review.
Cited by
-
Therapeutic potential of berberine in attenuating cholestatic liver injury: insights from a PSC mouse model.Cell Biosci. 2024 Jan 25;14(1):14. doi: 10.1186/s13578-024-01195-8. Cell Biosci. 2024. PMID: 38273376 Free PMC article.
-
Role of tryptophan-metabolizing microbiota in mice diarrhea caused by Folium sennae extracts.BMC Microbiol. 2020 Jun 29;20(1):185. doi: 10.1186/s12866-020-01864-x. BMC Microbiol. 2020. PMID: 32600333 Free PMC article.
-
Monoacylglycerol lipase reprograms lipid precursors signaling in liver disease.World J Gastroenterol. 2020 Jul 7;26(25):3577-3585. doi: 10.3748/wjg.v26.i25.3577. World J Gastroenterol. 2020. PMID: 32742127 Free PMC article. Review.
-
The Role of Metabolic Lipases in the Pathogenesis and Management of Liver Disease.Hepatology. 2020 Sep;72(3):1117-1126. doi: 10.1002/hep.31250. Hepatology. 2020. PMID: 32236963 Free PMC article. Review.
-
The Endocannabinoid System Drives Eosinophil Infiltration During Eosinophilic Esophagitis.Cell Mol Gastroenterol Hepatol. 2025;19(8):101515. doi: 10.1016/j.jcmgh.2025.101515. Epub 2025 Apr 11. Cell Mol Gastroenterol Hepatol. 2025. PMID: 40221089 Free PMC article.
References
-
- Dyson JK, Hirschfield GM, Adams DH, Beuers U, Mann DA, Lindor KD, et al. Novel therapeutic targets in primary biliary cirrhosis. Nat Rev Gastroenterol Hepatol 2015;12:147‐158. - PubMed
-
- Hirschfield GM, Heathcote EJ, Gershwin ME. Pathogenesis of cholestatic liver disease and therapeutic approaches. Gastroenterology 2010;139:1481‐1496. - PubMed
-
- Poursharifi P, Madiraju SRM, Prentki M. Monoacylglycerol signalling and ABHD6 in health and disease. Diabetes Obes Metab 2017;19:76‐89. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
