

Decoding the role of SOD2 in sickle cell disease
- PMID: 31506286
- PMCID: PMC6737422
- DOI: 10.1182/bloodadvances.2019000527
Decoding the role of SOD2 in sickle cell disease
Abstract
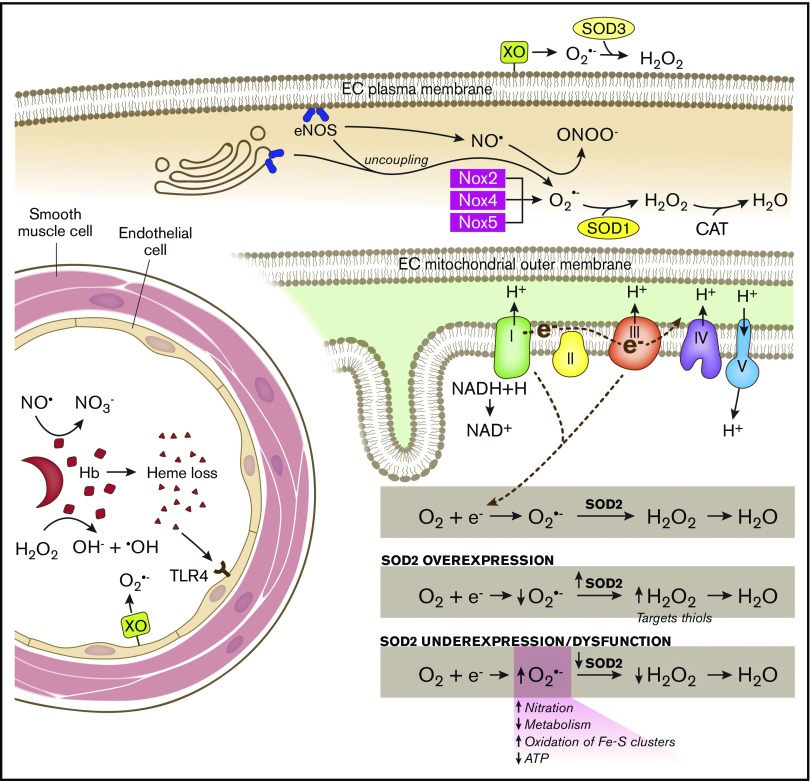
Sickle cell disease (SCD) is an inherited hemoglobinopathy caused by a single point mutation in the β-globin gene. As a consequence, deoxygenated hemoglobin polymerizes triggering red blood cell sickling and hemolysis, vaso-occlusion, and ischemia/reperfusion. Allied to these pathologies is the overproduction of reactive oxygen species driven by hemoglobin Fenton chemistry and peroxidase reactions as well as by secondary activation of vascular oxidases, including NAD(P)H oxidase and xanthine oxidase. In addition, hypoxia, produced by sickle red blood cell occlusion, disrupts mitochondrial metabolism and generates excess superoxide through electron leak from the mitochondrial respiratory chain. Superoxide dismutase 2 (SOD2) is a mitochondrial-specific antioxidant enzyme that dismutates superoxide to hydrogen peroxide, which is then converted to water by catalase and glutathione peroxidase. In SCD, the antioxidant defense system is significantly diminished through decreased expression and activity levels of antioxidant enzymes, including superoxide dismutase, catalase, and glutathione peroxidase. From a translational perspective, genetic variants including a missense variant in SOD2 (valine to alanine at position 16) are present in 45% of people with African ancestry and are associated with increased sickle complications. While it is known that there is an imbalance between oxidative species and antioxidant defenses in SCD, much more investigation is warranted. This review summarizes our current understanding of antioxidant defense systems in SCD, particularly focused on SOD2, and provides insight into challenges and opportunities as the field moves forward.
© 2019 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: A.C.S. receives research funding from Bayer Pharmaceuticals. The remaining authors declare no competing financial interests.
Figures
Similar articles
-
Reduced peripheral blood superoxide dismutase 2 expression in sickle cell disease.Ann Hematol. 2019 Jul;98(7):1561-1572. doi: 10.1007/s00277-019-03709-8. Epub 2019 May 16. Ann Hematol. 2019. PMID: 31098737 Clinical Trial.
-
SOD2 V16A amplifies vascular dysfunction in sickle cell patients by curtailing mitochondria complex IV activity.Blood. 2022 Mar 17;139(11):1760-1765. doi: 10.1182/blood.2021013350. Blood. 2022. PMID: 34958669 Free PMC article.
-
Does Deteriorating Antioxidant Defense and Impaired γ-Glutamyl Cycle Induce Oxidative Stress and Hemolysis in Individuals with Sickle Cell Disease?Antioxid Redox Signal. 2025 Feb;42(4-6):199-211. doi: 10.1089/ars.2024.0594. Epub 2024 Jul 29. Antioxid Redox Signal. 2025. PMID: 39001817
-
Innovations in Drug Discovery for Sickle Cell Disease Targeting Oxidative Stress and NRF2 Activation-A Short Review.Int J Mol Sci. 2025 Apr 28;26(9):4192. doi: 10.3390/ijms26094192. Int J Mol Sci. 2025. PMID: 40362428 Free PMC article. Review.
-
Impacts of oxidative stress and anti-oxidants on the development, pathogenesis, and therapy of sickle cell disease: A comprehensive review.Biomed Pharmacother. 2024 Jul;176:116849. doi: 10.1016/j.biopha.2024.116849. Epub 2024 Jun 1. Biomed Pharmacother. 2024. PMID: 38823275 Review.
Cited by
-
Gene Expression Profiling Elucidates Cellular Responses to NCX4040 in Human Ovarian Tumor Cells: Implications in the Mechanisms of Action of NCX4040.Cancers (Basel). 2022 Dec 31;15(1):285. doi: 10.3390/cancers15010285. Cancers (Basel). 2022. PMID: 36612280 Free PMC article.
-
Circular RNA circPHKA2 Relieves OGD-Induced Human Brain Microvascular Endothelial Cell Injuries through Competitively Binding miR-574-5p to Modulate SOD2.Oxid Med Cell Longev. 2021 Nov 8;2021:3823122. doi: 10.1155/2021/3823122. eCollection 2021. Oxid Med Cell Longev. 2021. PMID: 34790286 Free PMC article.
-
Redox Switches Controlling Nitric Oxide Signaling in the Resistance Vasculature and Implications for Blood Pressure Regulation: Mid-Career Award for Research Excellence 2020.Hypertension. 2021 Sep;78(4):912-926. doi: 10.1161/HYPERTENSIONAHA.121.16493. Epub 2021 Aug 23. Hypertension. 2021. PMID: 34420371 Free PMC article. Review.
-
Antioxidant capacity of fermented corn gluten meal in broiler chickens: a solid-state approach with mixed microbial fermentation.Poult Sci. 2024 Dec;103(12):104318. doi: 10.1016/j.psj.2024.104318. Epub 2024 Sep 12. Poult Sci. 2024. PMID: 39357236 Free PMC article.
-
Sickle Cell Disease Update: New Treatments and Challenging Nutritional Interventions.Nutrients. 2024 Jan 15;16(2):258. doi: 10.3390/nu16020258. Nutrients. 2024. PMID: 38257151 Free PMC article. Review.
References
-
- Ashley-Koch A, Yang Q, Olney RS. Sickle hemoglobin (HbS) allele and sickle cell disease: a HuGE review. Am J Epidemiol. 2000;151(9):839-845. - PubMed
-
- Chirico EN, Pialoux V. Role of oxidative stress in the pathogenesis of sickle cell disease. IUBMB Life. 2012;64(1):72-80. - PubMed
-
- Nur E, Biemond BJ, Otten HM, Brandjes DP, Schnog JJ; CURAMA Study Group . Oxidative stress in sickle cell disease; pathophysiology and potential implications for disease management. Am J Hematol. 2011;86(6):484-489. - PubMed
-
- Ware RE, de Montalembert M, Tshilolo L, Abboud MR. Sickle cell disease. Lancet. 2017;390(10091):311-323. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
