

Variant filtering, digenic variants, and other challenges in clinical sequencing: a lesson from fibrillinopathies
- PMID: 31506931
- PMCID: PMC7004123
- DOI: 10.1111/cge.13640
Variant filtering, digenic variants, and other challenges in clinical sequencing: a lesson from fibrillinopathies
Abstract
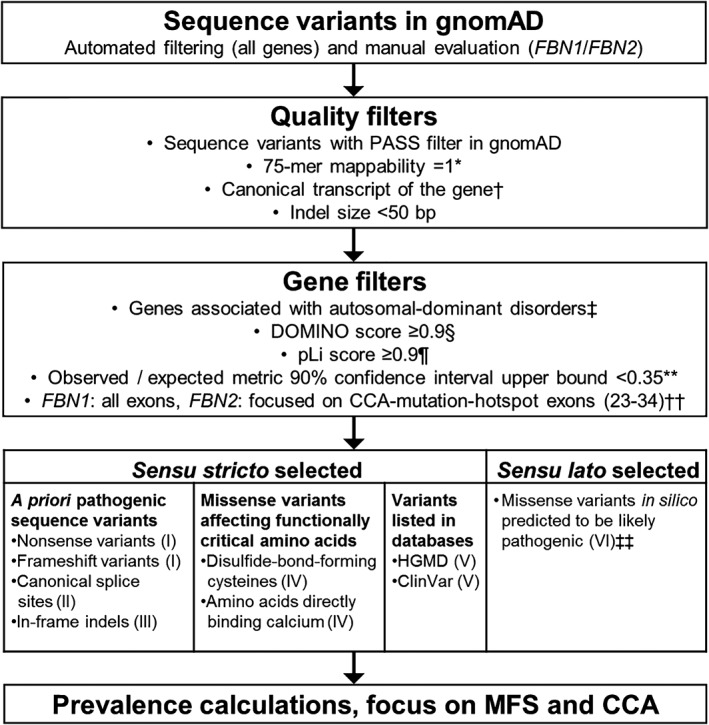
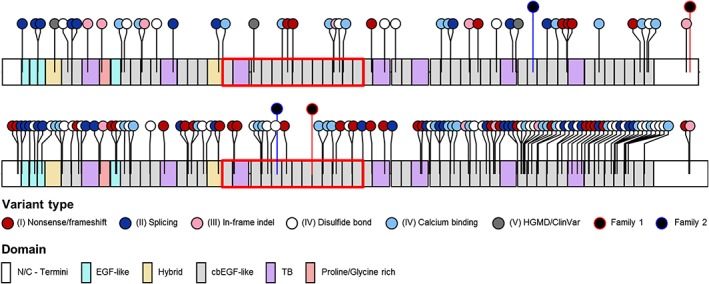
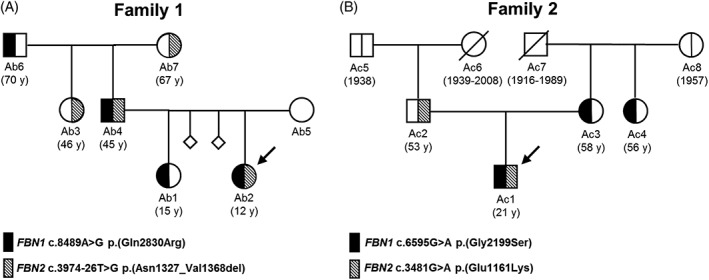
Genome-scale high-throughput sequencing enables the detection of unprecedented numbers of sequence variants. Variant filtering and interpretation are facilitated by mutation databases, in silico tools, and population-based reference datasets such as ExAC/gnomAD, while variants are classified using the ACMG/AMP guidelines. These methods, however, pose clinically relevant challenges. We queried the gnomAD dataset for (likely) pathogenic variants in genes causing autosomal-dominant disorders. Furthermore, focusing on the fibrillinopathies Marfan syndrome (MFS) and congenital contractural arachnodactyly (CCA), we screened 500 genomes of our patients for co-occurring variants in FBN1 and FBN2. In gnomAD, we detected 2653 (likely) pathogenic variants in 253 genes associated with autosomal-dominant disorders, enabling the estimation of variant-filtering thresholds and disease predisposition/prevalence rates. In our database, we discovered two families with hitherto unreported co-occurrence of FBN1/FBN2 variants causing phenotypes with mixed or modified MFS/CCA clinical features. We show that (likely) pathogenic gnomAD variants may be more frequent than expected and are challenging to classify according to the ACMG/AMP guidelines as well as that fibrillinopathies are likely underdiagnosed and may co-occur. Consequently, selection of appropriate frequency cutoffs, recognition of digenic variants, and variant classification represent considerable challenges in variant interpretation. Neglecting these challenges may lead to incomplete or missed diagnoses.
Keywords: Marfan syndrome; congenital contractural arachnodactyly; digenic variants; genome sequencing; variant interpretation.
© 2019 The Authors. Clinical Genetics published by John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Karczewski KJ, Francioli LC, Tiao G, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss‐of‐function intolerance across human protein‐coding genes. bioRxiv. 2019. 10.1101/531210. - DOI
-
- Bamshad MJ, Ng SB, Bigham AW, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12:745‐755. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
