

Metatranscriptomics yields new genomic resources and sensitive detection of infections for diverse blood parasites
- PMID: 31507097
- PMCID: PMC6942209
- DOI: 10.1111/1755-0998.13091
Metatranscriptomics yields new genomic resources and sensitive detection of infections for diverse blood parasites
Abstract
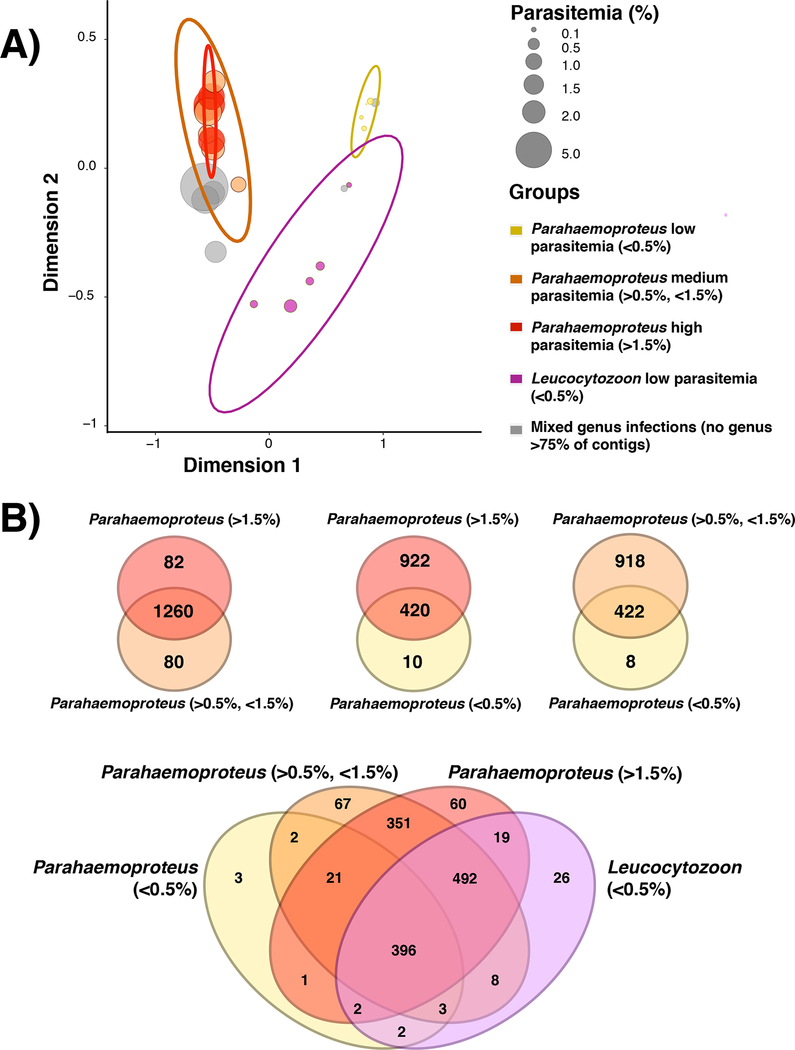
Metatranscriptomics is a powerful method for studying the composition and function of complex microbial communities. The application of metatranscriptomics to multispecies parasite infections is of particular interest, as research on parasite evolution and diversification has been hampered by technical challenges to genome-scale DNA sequencing. In particular, blood parasites of vertebrates are abundant and diverse although they often occur at low infection intensities and exist as multispecies infections, rendering the isolation of genomic sequence data challenging. Here, we use birds and their diverse haemosporidian parasites to illustrate the potential for metatranscriptome sequencing to generate large quantities of genome-wide sequence data from multiple blood parasite species simultaneously. We used RNA-sequencing of 24 blood samples from songbirds in North America to show that metatranscriptomes can yield large proportions of haemosporidian protein-coding gene repertoires even when infections are of low intensity (<0.1% red blood cells infected) and consist of multiple parasite taxa. By bioinformatically separating host and parasite transcripts and assigning them to the haemosporidian genus of origin, we found that transcriptomes detected ~23% more total parasite infections across all samples than were identified using microscopy and DNA barcoding. For single-species infections, we obtained data for >1,300 loci from samples with as low as 0.03% parasitaemia, with the number of loci increasing with infection intensity. In total, we provide data for 1,502 single-copy orthologous loci from a phylogenetically diverse set of 33 haemosporidian mitochondrial lineages. The metatranscriptomic approach described here has the potential to accelerate ecological and evolutionary research on haemosporidians and other diverse parasites.
Keywords: Leucocytozoon; Parahaemoproteus; Plasmodium; RNA-seq; co-infection; malaria parasite.
© 2019 John Wiley & Sons Ltd.
Figures



Similar articles
-
MOLECULAR SURVEY OF HAEMOSPORIDIAN PARASITES IN HAWKS, FALCONS, AND OWLS (ACCIPITRIFORMES, FALCONIFORMES, STRIGIFORMES) FROM MINNESOTA AND NORTH DAKOTA, WITH REMARKS ON THE PHYLOGENETIC RELATIONSHIPS OF HAEMOSPORIDIANS IN NORTH AMERICAN RAPTORS.J Parasitol. 2025 May 1;111(3):299-314. doi: 10.1645/25-16. J Parasitol. 2025. PMID: 40494546
-
Ecology, not distance, explains community composition in parasites of sky-island Audubon's Warblers.Int J Parasitol. 2019 May;49(6):437-448. doi: 10.1016/j.ijpara.2018.11.012. Epub 2019 Mar 23. Int J Parasitol. 2019. PMID: 30910465
-
Haemosporidioses in wild Eurasian blackbirds (Turdus merula) and song thrushes (T. philomelos): an in situ hybridization study with emphasis on exo-erythrocytic parasite burden.Malar J. 2020 Feb 12;19(1):69. doi: 10.1186/s12936-020-3147-6. Malar J. 2020. PMID: 32050970 Free PMC article.
-
Diptera vectors of avian Haemosporidian parasites: untangling parasite life cycles and their taxonomy.Biol Rev Camb Philos Soc. 2012 Nov;87(4):928-64. doi: 10.1111/j.1469-185X.2012.00234.x. Epub 2012 May 23. Biol Rev Camb Philos Soc. 2012. PMID: 22616880 Review.
-
Origin and diversity of malaria parasites and other Haemosporida.Trends Parasitol. 2023 Jul;39(7):501-516. doi: 10.1016/j.pt.2023.04.004. Epub 2023 May 16. Trends Parasitol. 2023. PMID: 37202254 Review.
Cited by
-
Contrasting drivers of diversity in hosts and parasites across the tropical Andes.Proc Natl Acad Sci U S A. 2021 Mar 23;118(12):e2010714118. doi: 10.1073/pnas.2010714118. Proc Natl Acad Sci U S A. 2021. PMID: 33731475 Free PMC article.
-
Whole blood transcriptomics analysis of Indonesians reveals translocated and pathogenic microbiota in blood.PLoS One. 2025 Jul 24;20(7):e0328788. doi: 10.1371/journal.pone.0328788. eCollection 2025. PLoS One. 2025. PMID: 40705807 Free PMC article.
-
Prevalence and Diversity of Haemosporidian-Associated Matryoshka RNA Viruses in a Natural Population of Wild Birds.Ecol Evol. 2025 May 26;15(5):e71239. doi: 10.1002/ece3.71239. eCollection 2025 May. Ecol Evol. 2025. PMID: 40421060 Free PMC article.
-
Vive la Différence: Exploiting the Differences between Rodent and Human Malarias.Trends Parasitol. 2020 Jun;36(6):504-511. doi: 10.1016/j.pt.2020.03.008. Epub 2020 Apr 16. Trends Parasitol. 2020. PMID: 32407681 Free PMC article. Review.
-
Novel RNA viruses associated with avian haemosporidian parasites.PLoS One. 2022 Jun 30;17(6):e0269881. doi: 10.1371/journal.pone.0269881. eCollection 2022. PLoS One. 2022. PMID: 35771829 Free PMC article.
References
-
- Andrews S (2010). FastQC: a quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
