

Repertoire Sequencing of B Cells Elucidates the Role of UNG and Mismatch Repair Proteins in Somatic Hypermutation in Humans
- PMID: 31507588
- PMCID: PMC6718458
- DOI: 10.3389/fimmu.2019.01913
Repertoire Sequencing of B Cells Elucidates the Role of UNG and Mismatch Repair Proteins in Somatic Hypermutation in Humans
Abstract
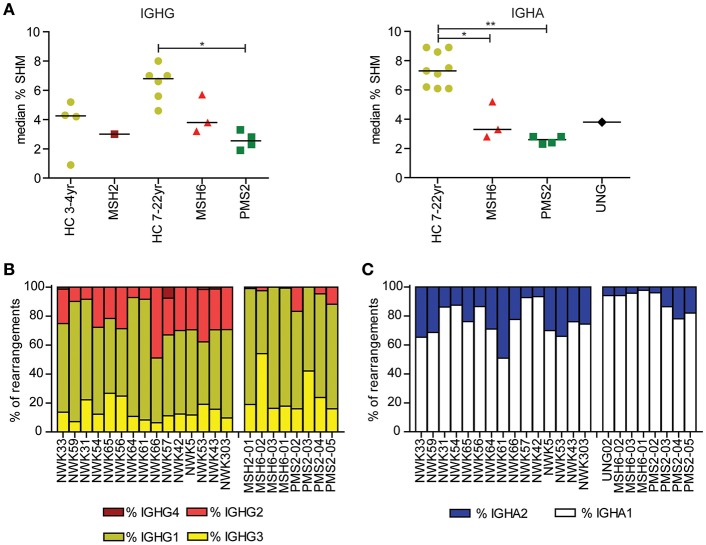
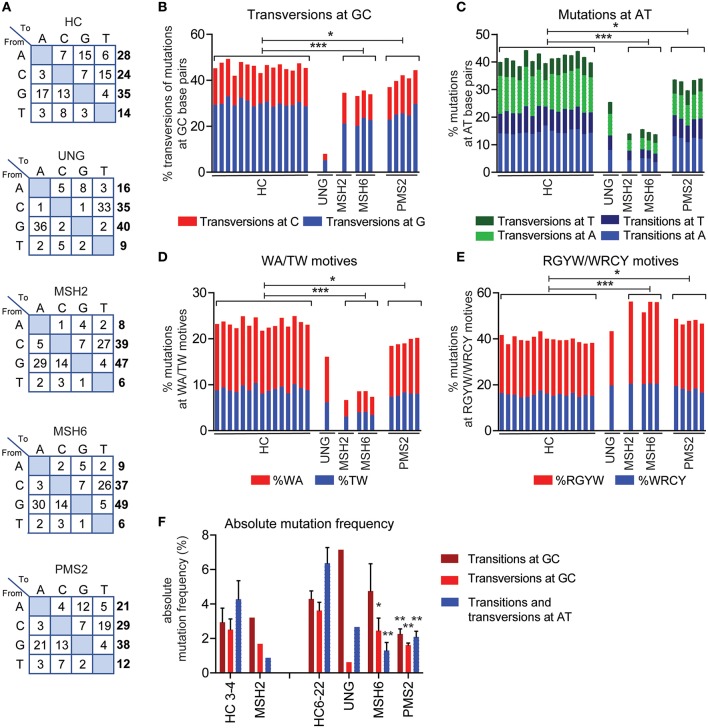
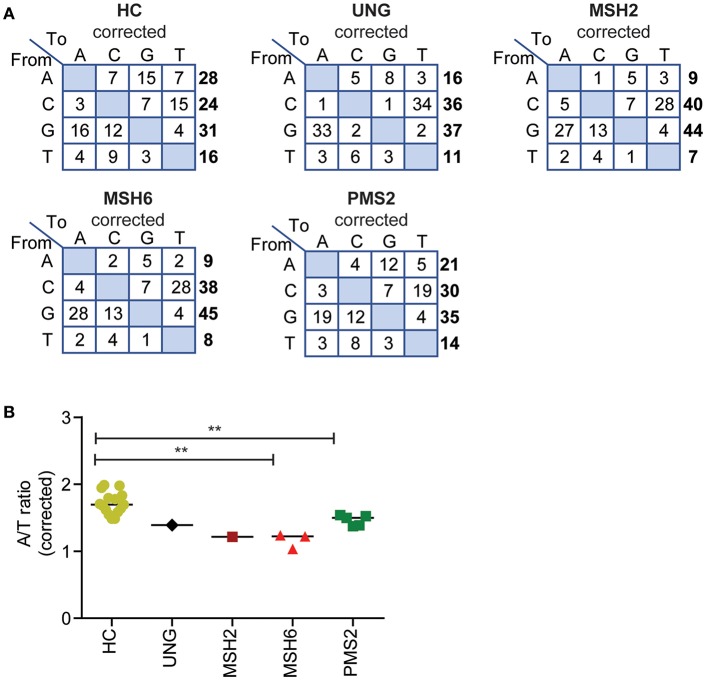
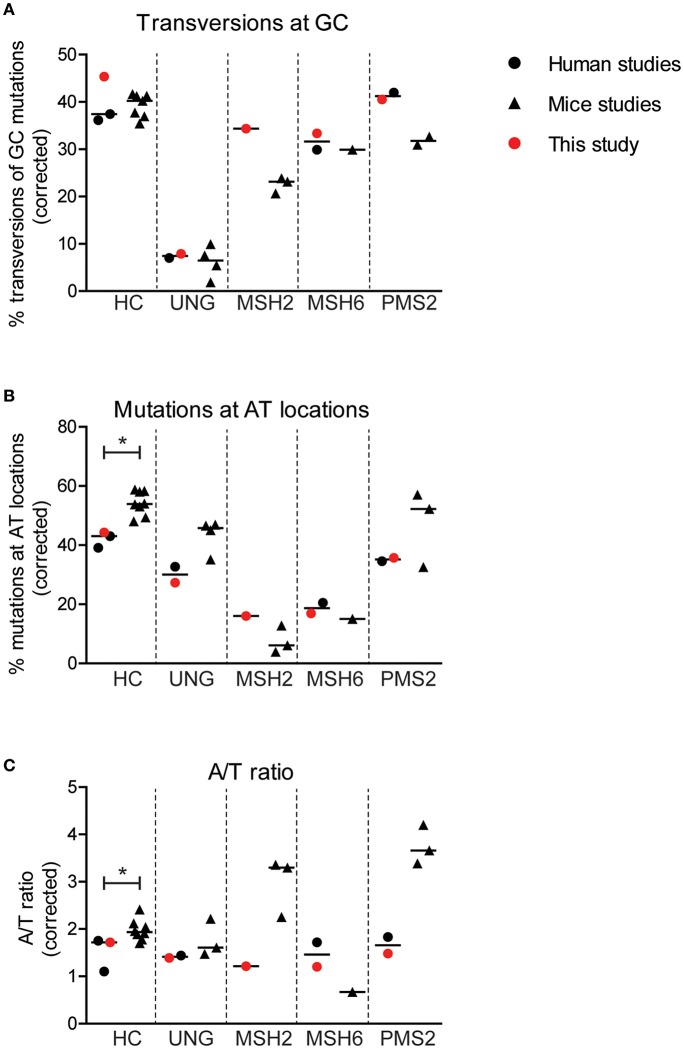
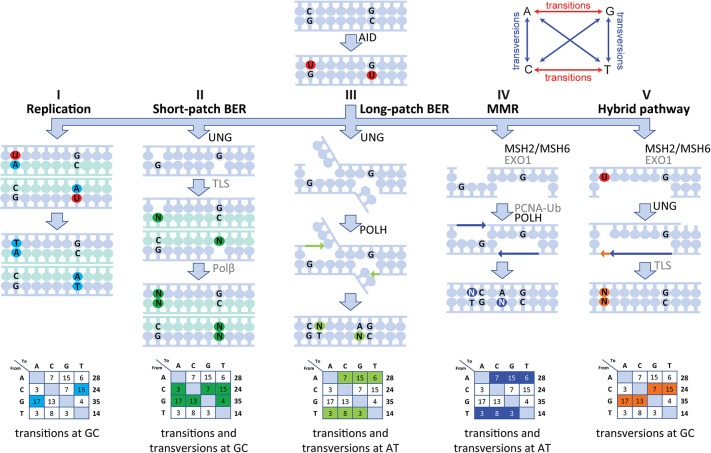
The generation of high-affinity antibodies depends on somatic hypermutation (SHM). SHM is initiated by the activation-induced cytidine deaminase (AID), which generates uracil (U) lesions in the B-cell receptor (BCR) encoding genes. Error-prone processing of U lesions creates a typical spectrum of point mutations during SHM. The aim of this study was to determine the molecular mechanism of SHM in humans; currently available knowledge is limited by the number of mutations analyzed per patient. We collected a unique cohort of 10 well-defined patients with bi-allelic mutations in genes involved in base excision repair (BER) (UNG) or mismatch repair (MMR) (MSH2, MSH6, or PMS2) and are the first to present next-generation sequencing (NGS) data of the BCR, allowing us to study SHM extensively in humans. Analysis using ARGalaxy revealed selective skewing of SHM mutation patterns specific for each genetic defect, which are in line with the five-pathway model of SHM that was recently proposed based on mice data. However, trans-species comparison revealed differences in the role of PMS2 and MSH2 in strand targeting between mice and man. In conclusion, our results indicate a role for UNG, MSH2, MSH6, and PMS2 in the generation of SHM in humans comparable to their function in mice. However, we observed differences in strand targeting between humans and mice, emphasizing the importance of studying molecular mechanisms in a human setting. The here developed method combining NGS and ARGalaxy analysis of BCR mutation data forms the basis for efficient SHM analyses of other immune deficiencies.
Keywords: B cells; B-cell receptor; DNA repair; base excision repair (BER); constitutional mismatch repair deficiency (CMMRD); immunoglobulin; mismatch repair (MMR); somatic hypermutation.
Figures
Similar articles
-
Base Excision Repair in the Immune System: Small DNA Lesions With Big Consequences.Front Immunol. 2020 May 29;11:1084. doi: 10.3389/fimmu.2020.01084. eCollection 2020. Front Immunol. 2020. PMID: 32547565 Free PMC article. Review.
-
DNA polymerases β and λ do not directly affect Ig variable region somatic hypermutation although their absence reduces the frequency of mutations.DNA Repair (Amst). 2013 Dec;12(12):1087-93. doi: 10.1016/j.dnarep.2013.09.002. Epub 2013 Sep 29. DNA Repair (Amst). 2013. PMID: 24084171 Free PMC article.
-
Somatic hypermutation and class switch recombination in Msh6(-/-)Ung(-/-) double-knockout mice.J Immunol. 2006 Oct 15;177(8):5386-92. doi: 10.4049/jimmunol.177.8.5386. J Immunol. 2006. PMID: 17015724
-
Differential expression of APE1 and APE2 in germinal centers promotes error-prone repair and A:T mutations during somatic hypermutation.Proc Natl Acad Sci U S A. 2014 Jun 24;111(25):9217-22. doi: 10.1073/pnas.1405590111. Epub 2014 Jun 9. Proc Natl Acad Sci U S A. 2014. PMID: 24927551 Free PMC article.
-
Mutating for Good: DNA Damage Responses During Somatic Hypermutation.Front Immunol. 2019 Mar 12;10:438. doi: 10.3389/fimmu.2019.00438. eCollection 2019. Front Immunol. 2019. PMID: 30915081 Free PMC article. Review.
Cited by
-
Tandem Substitutions in Somatic Hypermutation.Front Immunol. 2022 Jan 7;12:807015. doi: 10.3389/fimmu.2021.807015. eCollection 2021. Front Immunol. 2022. PMID: 35069591 Free PMC article.
-
Base Excision Repair in the Immune System: Small DNA Lesions With Big Consequences.Front Immunol. 2020 May 29;11:1084. doi: 10.3389/fimmu.2020.01084. eCollection 2020. Front Immunol. 2020. PMID: 32547565 Free PMC article. Review.
-
NGS-Based B-Cell Receptor Repertoire AnalysisRepertoire analyses in the Context of Inborn Errors of Immunity.Methods Mol Biol. 2022;2453:169-190. doi: 10.1007/978-1-0716-2115-8_11. Methods Mol Biol. 2022. PMID: 35622327 Free PMC article.
-
Position-Dependent Differential Targeting of Somatic Hypermutation.J Immunol. 2020 Dec 15;205(12):3468-3479. doi: 10.4049/jimmunol.2000496. Epub 2020 Nov 13. J Immunol. 2020. PMID: 33188076 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous