

Interleukin-1 Family Cytokines: Keystones in Liver Inflammatory Diseases
- PMID: 31507607
- PMCID: PMC6718562
- DOI: 10.3389/fimmu.2019.02014
Interleukin-1 Family Cytokines: Keystones in Liver Inflammatory Diseases
Abstract
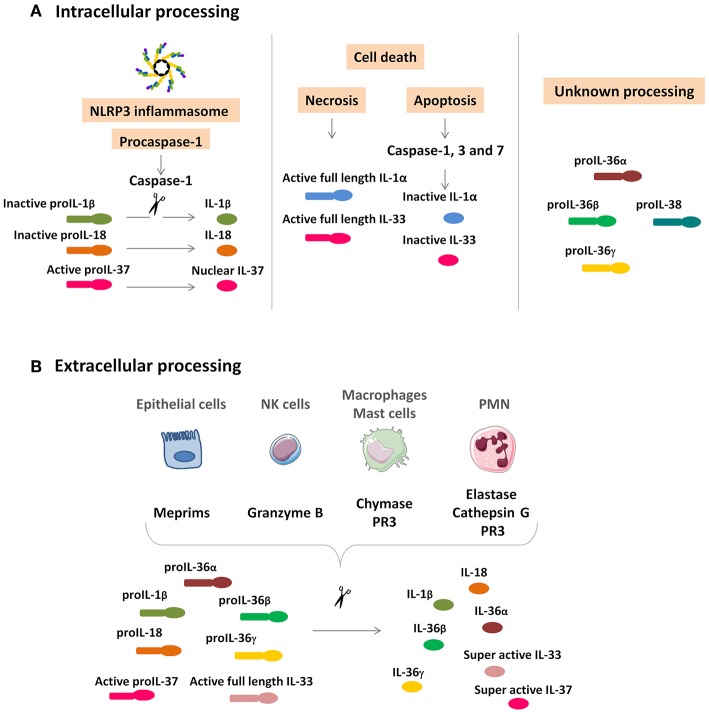
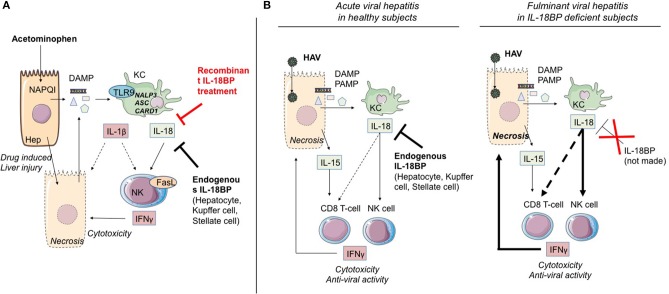
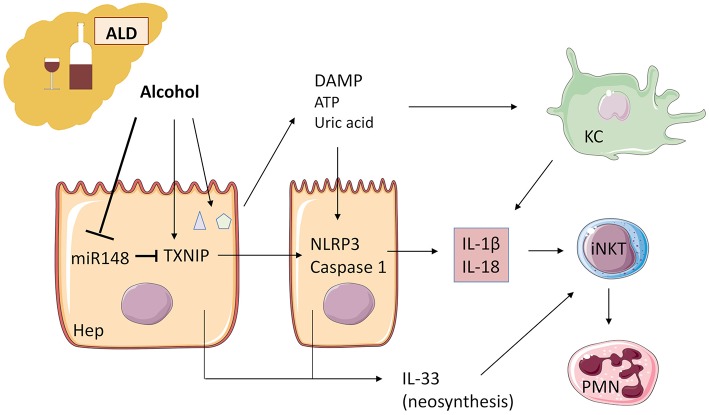
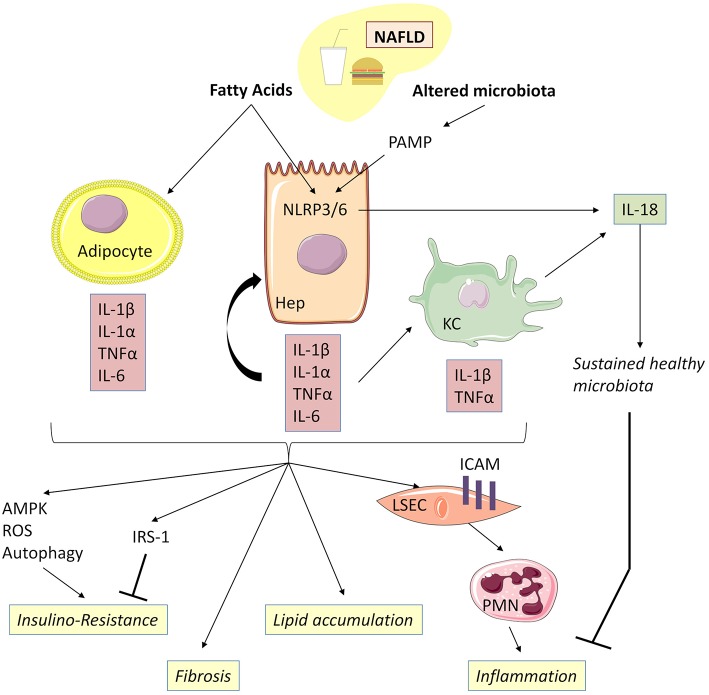
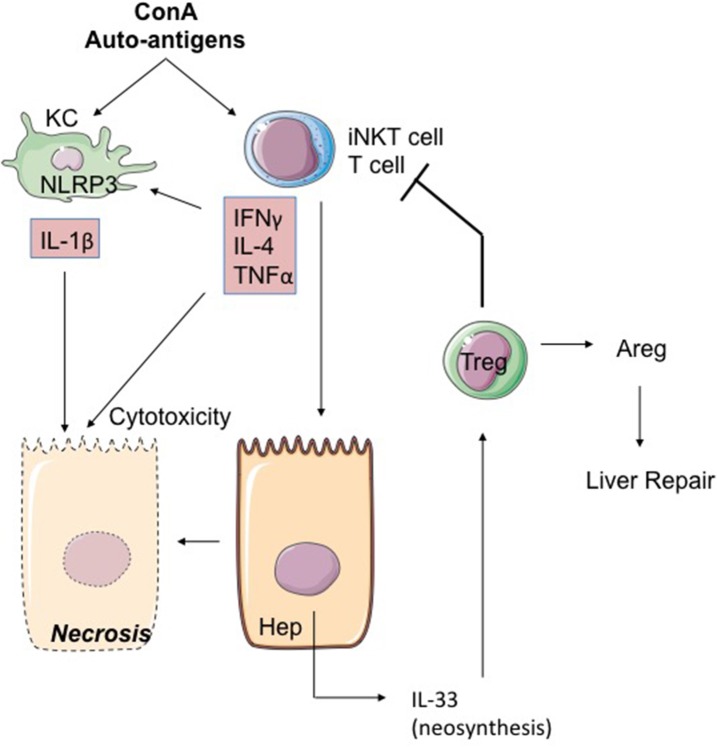
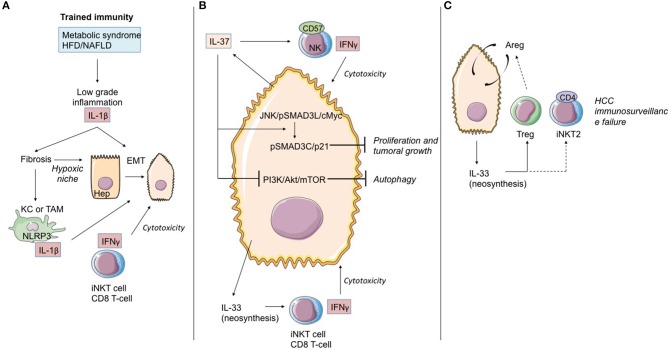
The pyrogenic property being the first activity described, members of the interleukin-1 superfamily (IL-1α, IL-1β, IL-18, and the newest members: IL-33, IL-36, IL-37, and IL-38) are now known to be involved in several inflammatory diseases such as obesity, atherosclerosis, cancer, viral and parasite infections, and auto-inflammatory syndromes as well as liver diseases. Inflammation processes are keystones of chronic liver diseases, of which the etiology may be viral or toxic, as in alcoholic or non-alcoholic liver diseases. Inflammation is also at stake in acute liver failure involving massive necrosis, and in ischemia-reperfusion injury in the setting of liver transplantation. The role of the IL-1 superfamily of cytokines and receptors in liver diseases can be either protective or pro-inflammatory, depending on timing and the environment. Our review provides an overview of current understanding of the IL-1 family members in liver inflammation, highlighting recent key investigations, and therapeutic perspectives. We have tried to apply the concept of trained immunity to liver diseases, based on the role of the members of the IL-1 superfamily, first of all IL-1β but also IL-18 and IL-33, in modulating innate lymphoid immunity carried by natural killer cells, innate lymphoid cells or innate T-αβ lymphocytes.
Keywords: inflammation; innate immunity; innate lymphoid cells; interleukin-1 family cytokines; invariant natural killer T-cells; liver diseases; natural killer cells; trained immunity.
Figures
References
-
- Beeson PB. Temperature-elevating effect of a substance obtained from polymorphonuclear leucocytes. J Clin Invest. (1948) 27:524. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous