

Xenografting of human umbilical mesenchymal stem cells from Wharton's jelly ameliorates mouse spinocerebellar ataxia type 1
- PMID: 31508229
- PMCID: PMC6727337
- DOI: 10.1186/s40035-019-0166-8
Xenografting of human umbilical mesenchymal stem cells from Wharton's jelly ameliorates mouse spinocerebellar ataxia type 1
Abstract
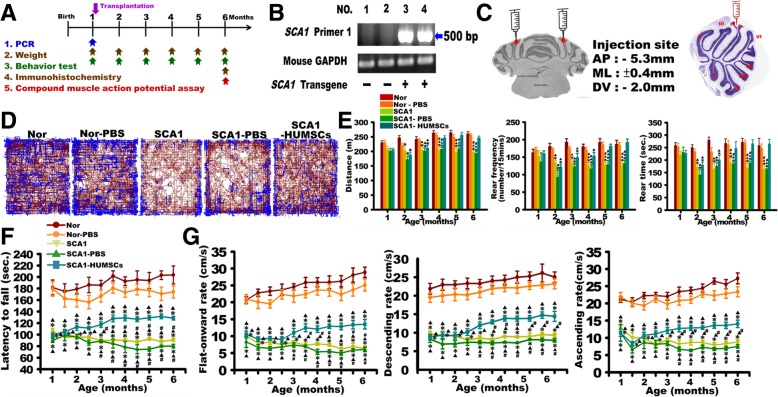
Background: Spinocerebellar ataxia type 1 (SCA1) is an autosomal dominant neurodegenerative disorder caused by the expansion of CAG repeats in ATXN1 gene resulting in an expansion of polyglutamine repeats in the ATXN1 protein. Unfortunately, there has yet been any effective treatment so far for SCA1. This study investigated the feasibility of transplanting human umbilical mesenchymal stem cells (HUMSCs) into transgenic SCA1 mice containing an expanded uninterrupted allele with 82 repeats in the ATXN1-coding region.
Methods: 106 human umbilical mesenchymal stem cells were transplanted into the cerebella at 1 month of age.
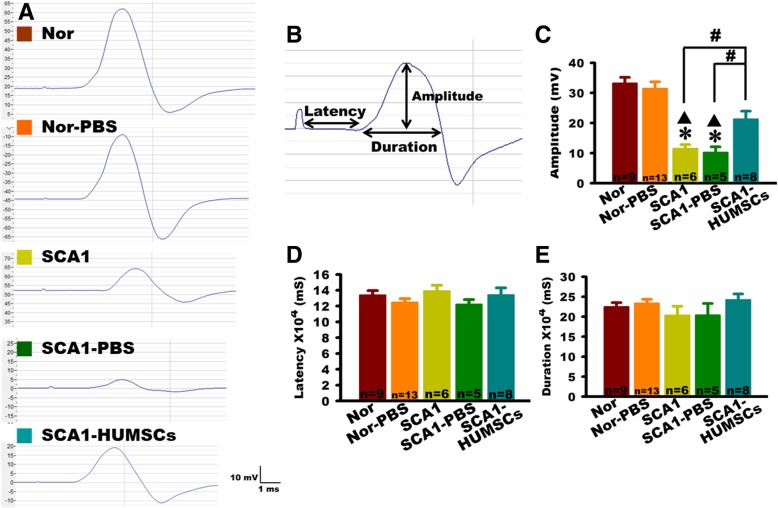
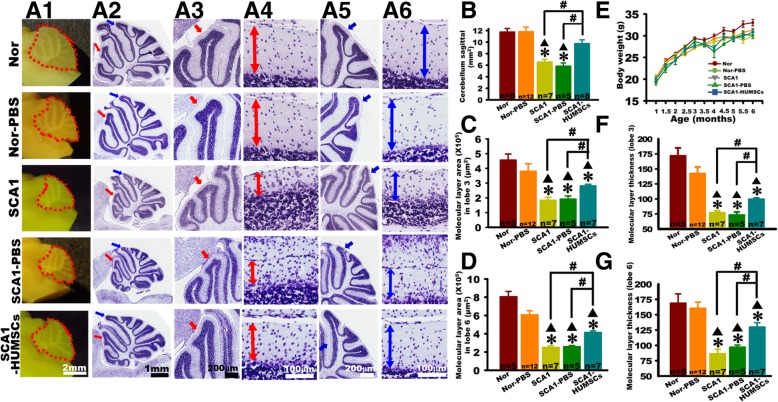
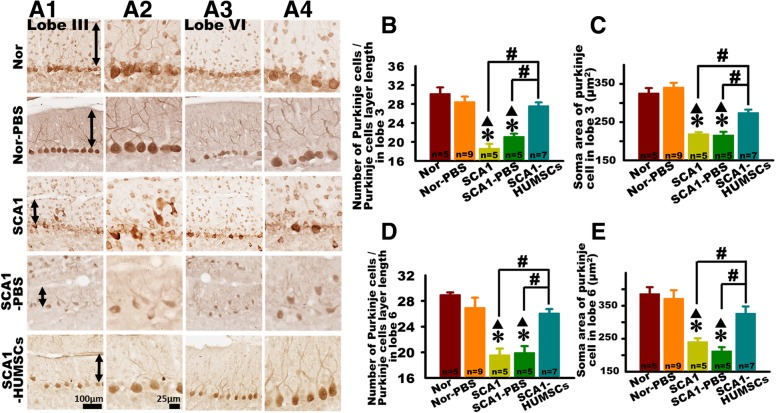
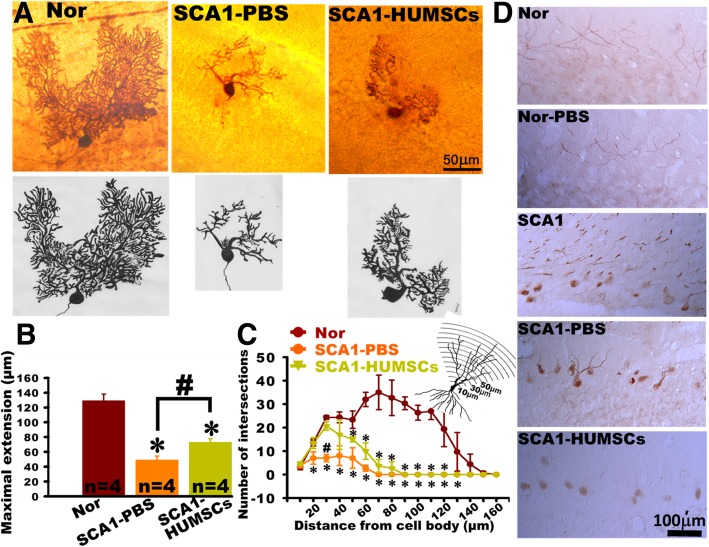
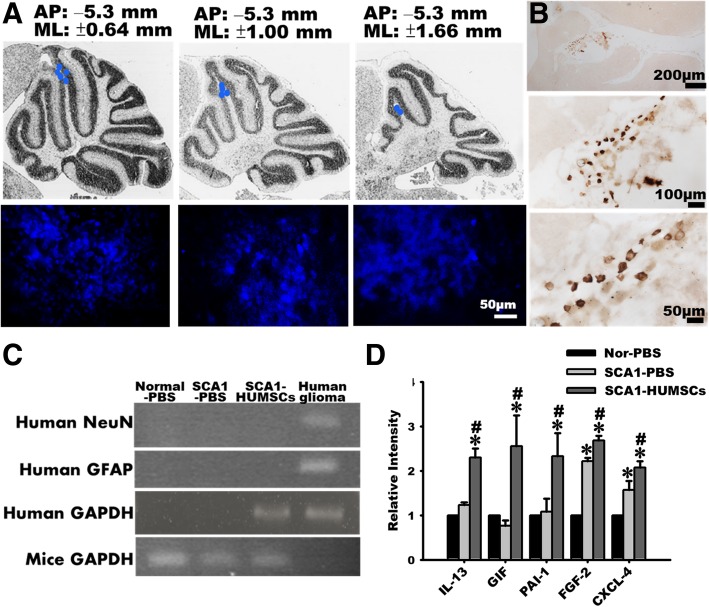
Results: HUMSCs displayed significant ameliorating effects in SCA1 mice in terms of motor behaviors in balance beam test and open field test as compared with the untransplanted SCA1 mice. HUMSCs transplantation effectively reduced the cerebellar atrophy, salvaged Purkinje cell death, and alleviated molecular layer shrinkage. Electrophysiological studies showed higher amplitudes of compound motor action potentials as indicated by increasing neuronal-muscular response strength to stimuli after stem cell transplantation. At 5 months after transplantation, HUMSCs scattering in the mice cerebella remained viable and secreted cytokines without differentiating into neuronal or glia cells.
Conclusions: Our findings provide hope for a new therapeutic direction for the treatment of SCA1.
Keywords: Cell transplantation; SCA1; Umbilical mesenchymal stem cells.
Conflict of interest statement
Competing interestsThe authors declare that they have no competing interests.
Figures
References
-
- Soong BW, Morrison PJ. The cerebellum disorders and treatment. In: Manto M, Huisman TAGM, editors. Handbook of clinical neurology, Vol. 155 (3rd series), Chapter 10: Spinocerebellar ataxias. Amsterdam: Elsevier; 2018. p. 143–74. - PubMed
-
- Banfi S, Chung MY, Kwiatkowski TJ, Jr, Ranum LP, McCall AE, Chinault AC, Orr HT, Zoghbi HY. Mapping and cloning of the critical region for the spinocerebellar ataxia type 1 gene (SCA1) in a yeast artificial chromosome contig spanning 1.2 Mb. Genomics. 1993;18(3):627–635. doi: 10.1016/S0888-7543(05)80365-9. - DOI - PubMed
LinkOut - more resources
Full Text Sources
