

Baseline human gut microbiota profile in healthy people and standard reporting template
- PMID: 31509535
- PMCID: PMC6738582
- DOI: 10.1371/journal.pone.0206484
Baseline human gut microbiota profile in healthy people and standard reporting template
Abstract
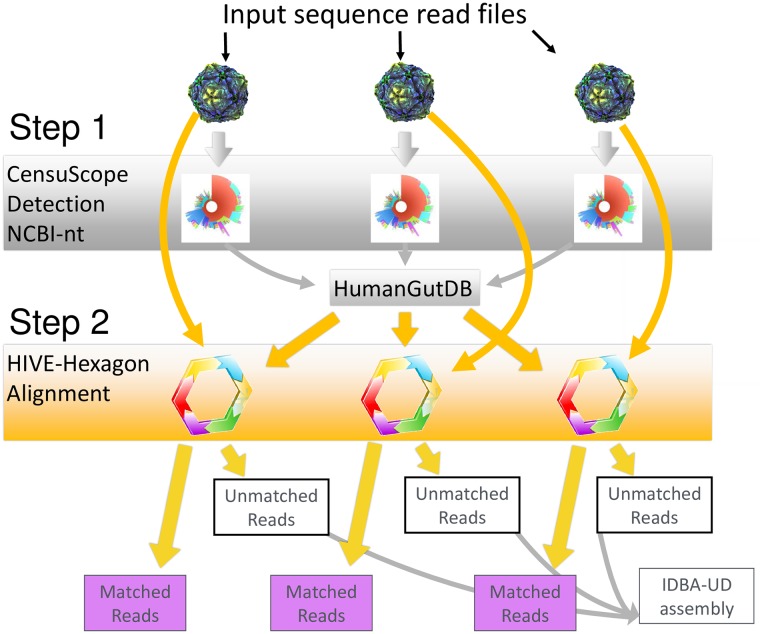
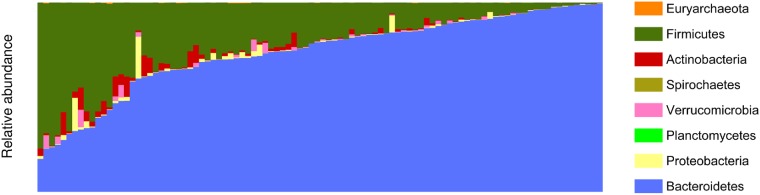
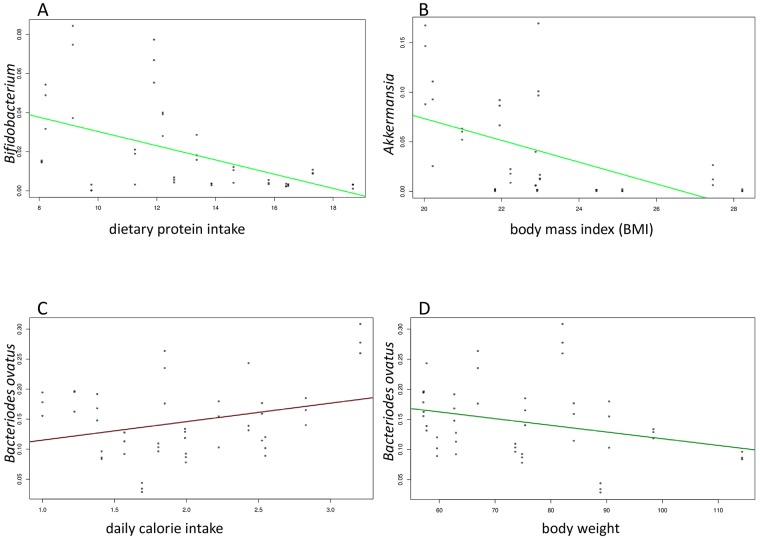
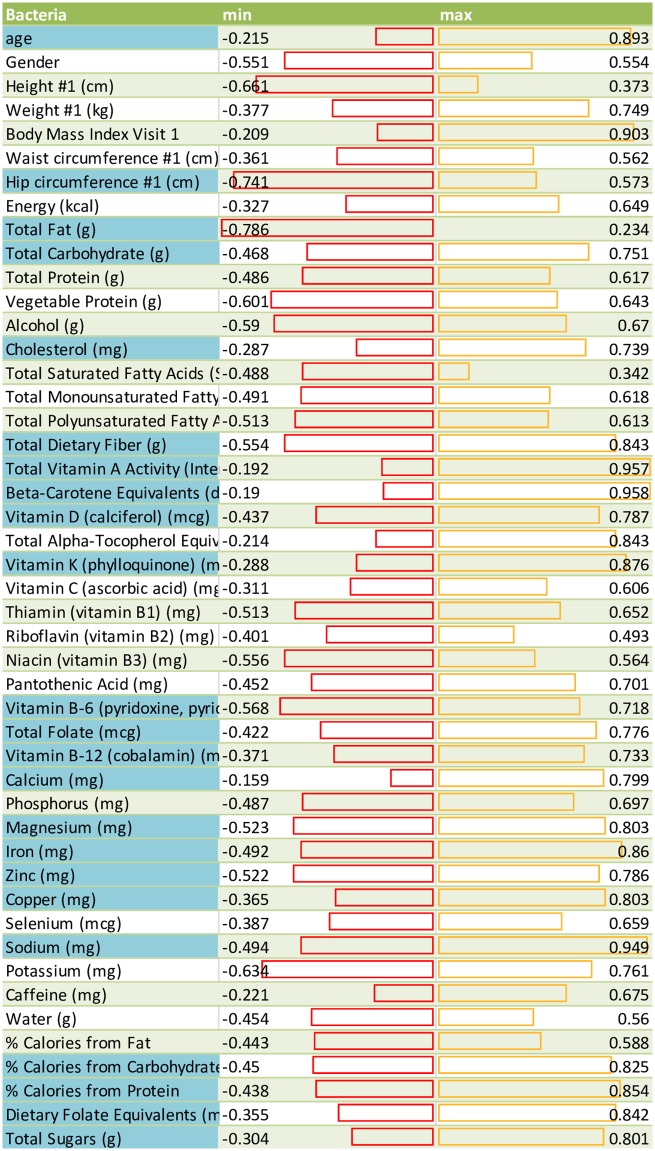
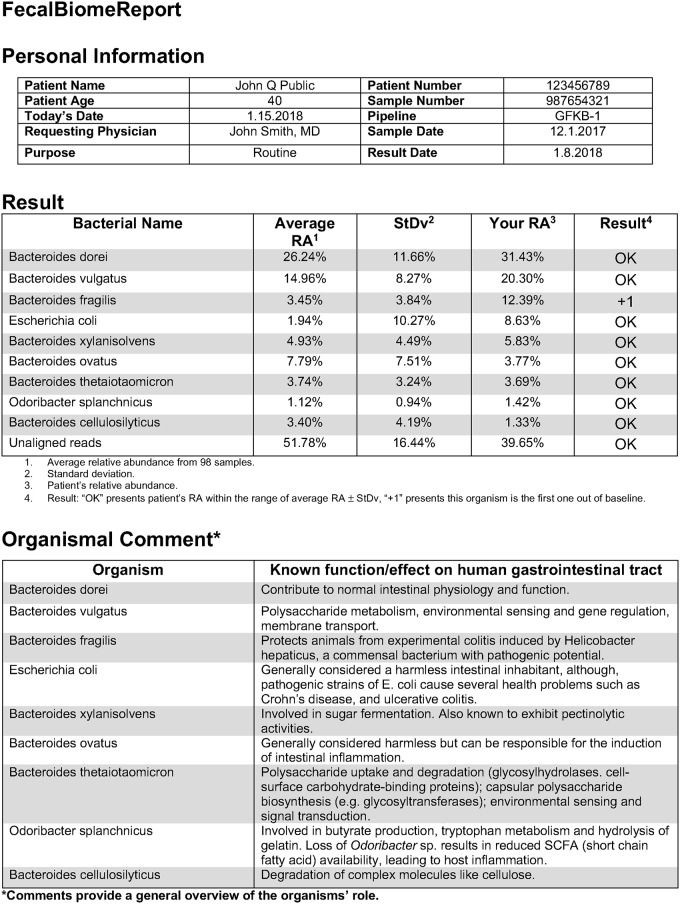
A comprehensive knowledge of the types and ratios of microbes that inhabit the healthy human gut is necessary before any kind of pre-clinical or clinical study can be performed that attempts to alter the microbiome to treat a condition or improve therapy outcome. To address this need we present an innovative scalable comprehensive analysis workflow, a healthy human reference microbiome list and abundance profile (GutFeelingKB), and a novel Fecal Biome Population Report (FecalBiome) with clinical applicability. GutFeelingKB provides a list of 157 organisms (8 phyla, 18 classes, 23 orders, 38 families, 59 genera and 109 species) that forms the baseline biome and therefore can be used as healthy controls for studies related to dysbiosis. This list can be expanded to 863 organisms if closely related proteomes are considered. The incorporation of microbiome science into routine clinical practice necessitates a standard report for comparison of an individual's microbiome to the growing knowledgebase of "normal" microbiome data. The FecalBiome and the underlying technology of GutFeelingKB address this need. The knowledgebase can be useful to regulatory agencies for the assessment of fecal transplant and other microbiome products, as it contains a list of organisms from healthy individuals. In addition to the list of organisms and their abundances, this study also generated a collection of assembled contiguous sequences (contigs) of metagenomics dark matter. In this study, metagenomic dark matter represents sequences that cannot be mapped to any known sequence but can be assembled into contigs of 10,000 nucleotides or higher. These sequences can be used to create primers to study potential novel organisms. All data is freely available from https://hive.biochemistry.gwu.edu/gfkb and NCBI's Short Read Archive.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
