

A Unique Epigenomic Landscape Defines Human Erythropoiesis
- PMID: 31509757
- PMCID: PMC6863094
- DOI: 10.1016/j.celrep.2019.08.020
A Unique Epigenomic Landscape Defines Human Erythropoiesis
Abstract
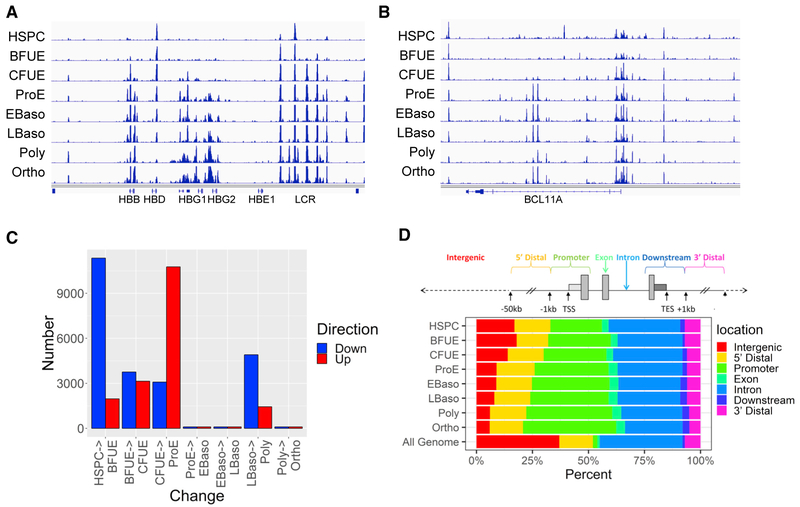
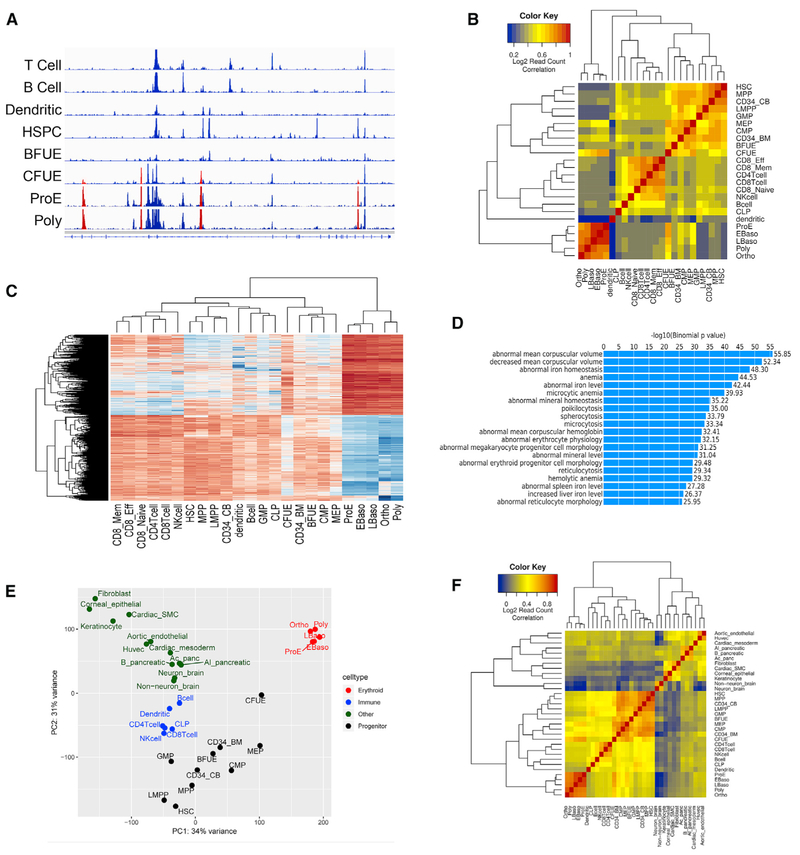
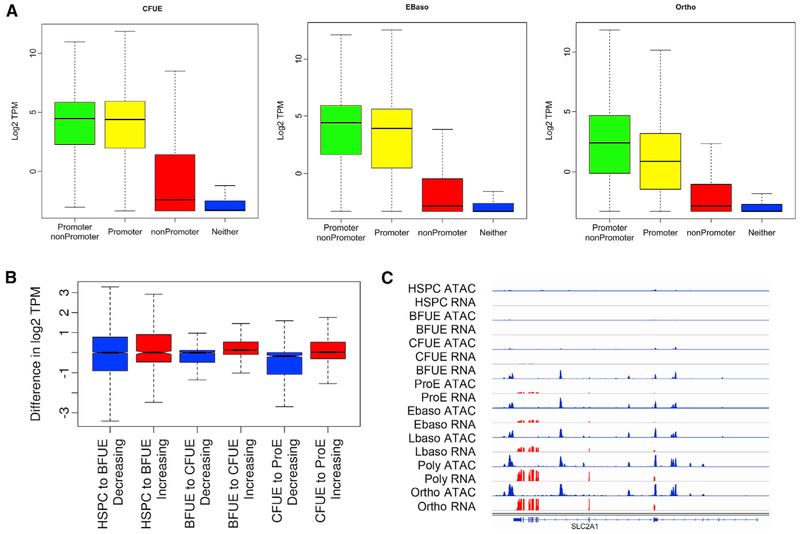
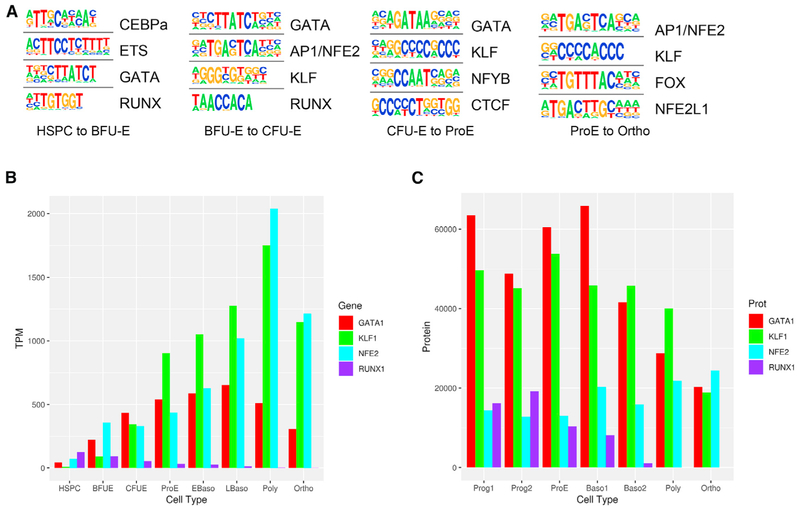
Mammalian erythropoiesis yields a highly specialized cell type, the mature erythrocyte, evolved to meet the organismal needs of increased oxygen-carrying capacity. To better understand the regulation of erythropoiesis, we performed genome-wide studies of chromatin accessibility, DNA methylation, and transcriptomics using a recently developed strategy to obtain highly purified populations of primary human erythroid cells. The integration of gene expression, DNA methylation, and chromatin state dynamics reveals that stage-specific gene regulation during erythropoiesis is a stepwise and hierarchical process involving many cis-regulatory elements. Erythroid-specific, nonpromoter sites of chromatin accessibility are linked to erythroid cell phenotypic variation and inherited disease. Comparative analyses of stage-specific chromatin accessibility indicate that there is limited early chromatin priming of erythroid genes during hematopoiesis. The epigenome of terminally differentiating erythroid cells defines a distinct subset of highly specialized cells that are vastly dissimilar from other hematopoietic and nonhematopoietic cell types. These epigenomic and transcriptome data are powerful tools to study human erythropoiesis.
Keywords: anemia; chromatin; epigenomic; erythrocyte; erythropoiesis; methylation; trait.
Copyright © 2019 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
The authors declare no competing interests.
Figures
References
-
- Akalin A, Garrett-Bakelman FE, Kormaksson M, Busuttil J, Zhang L, Khrebtukova I, Milne TA, Huang Y, Biswas D, Hess JL, et al. (2012a). Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet. 8, e1002781. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
