

NAD hydrolysis by the tuberculosis necrotizing toxin induces lethal oxidative stress in macrophages
- PMID: 31509891
- PMCID: PMC6925628
- DOI: 10.1111/cmi.13115
NAD hydrolysis by the tuberculosis necrotizing toxin induces lethal oxidative stress in macrophages
Abstract
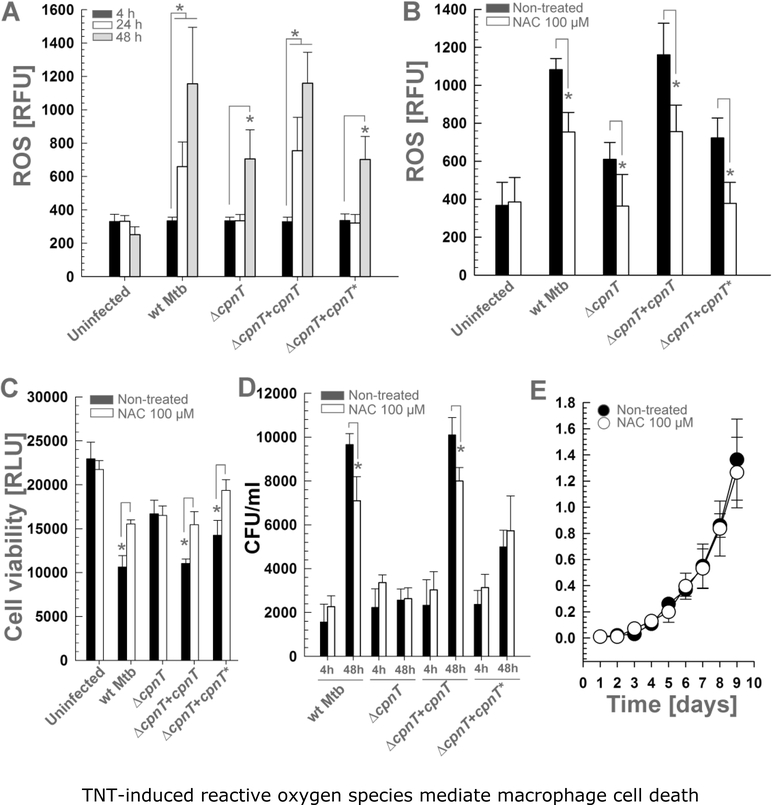
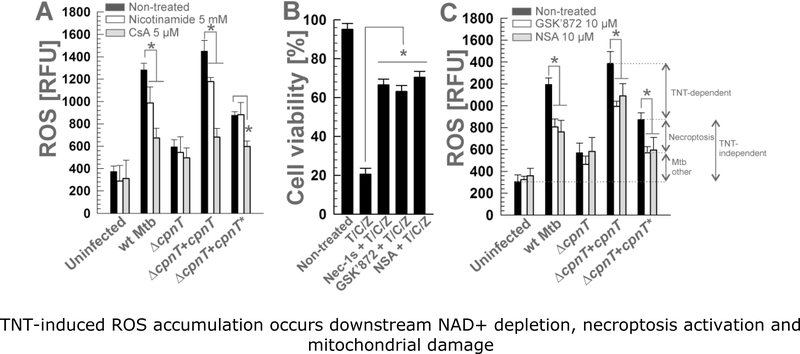
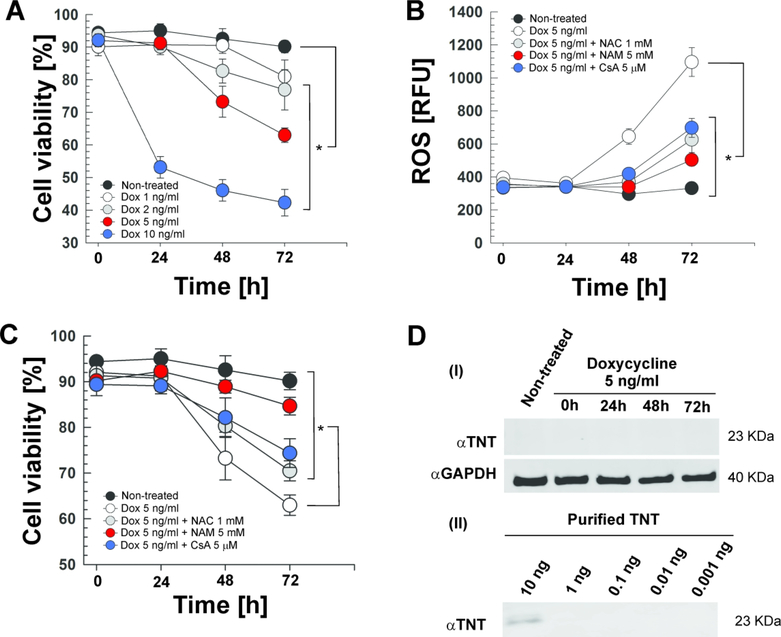
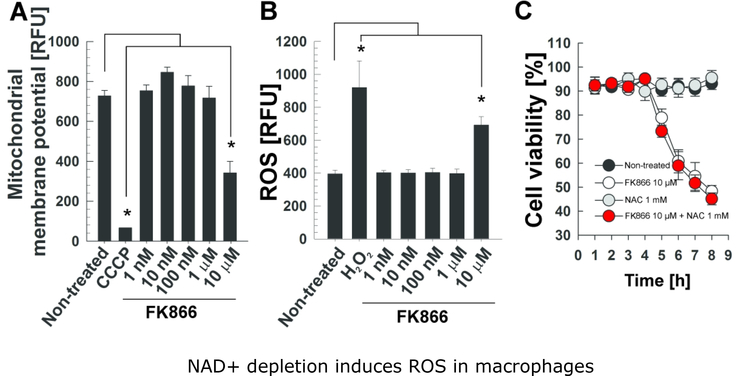
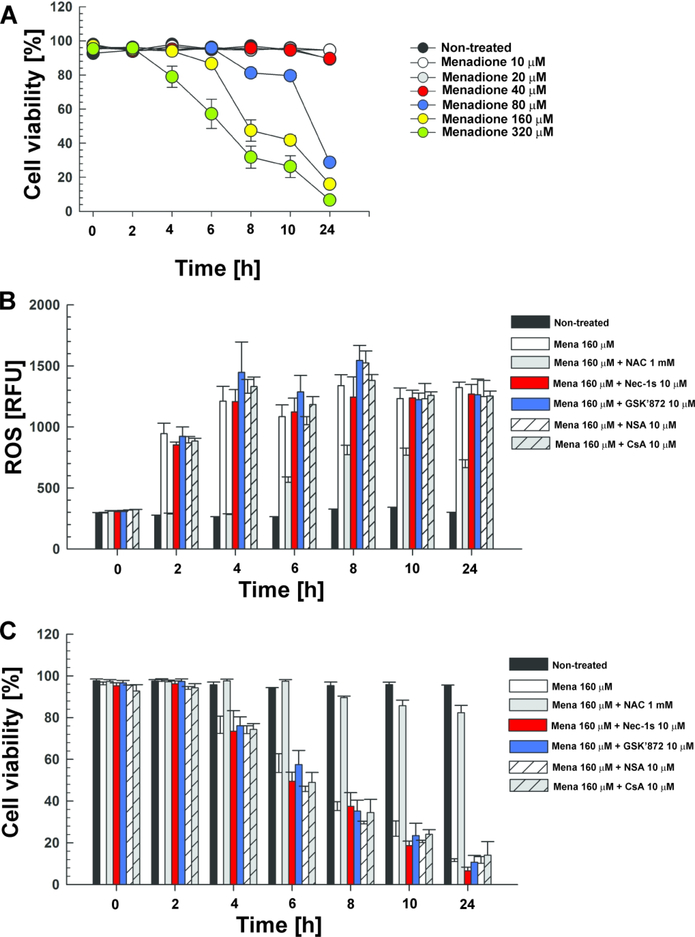
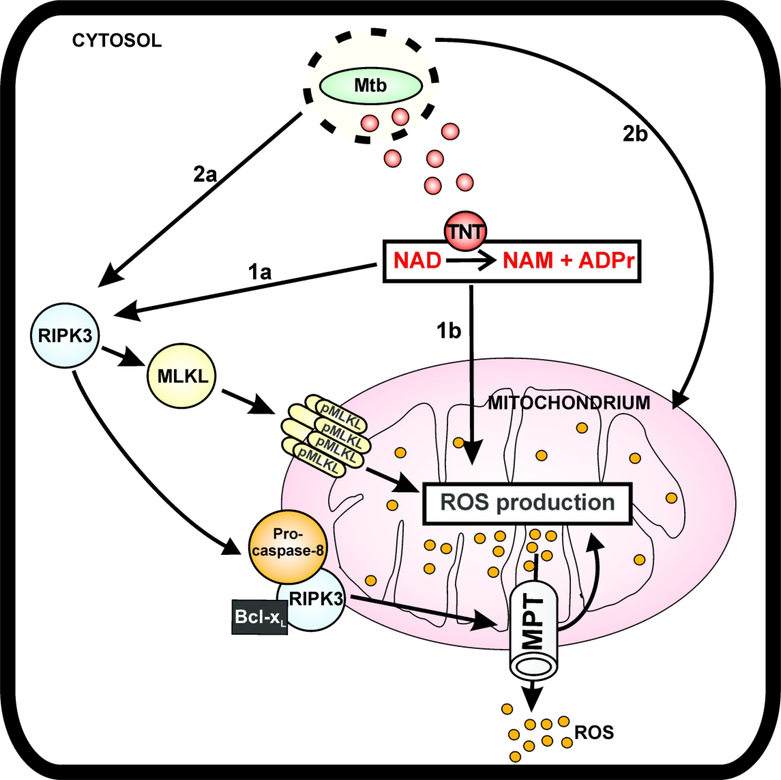
Mycobacterium tuberculosis (Mtb) kills infected macrophages through necroptosis, a programmed cell death that enhances mycobacterial replication and dissemination. The tuberculosis necrotizing toxin (TNT) is the major cytotoxicity factor of Mtb in macrophages and induces necroptosis by NAD+ hydrolysis. Here, we show that the catalytic activity of TNT triggers the production of reactive oxygen species (ROS) in Mtb-infected macrophages causing cell death and promoting mycobacterial replication. TNT induces ROS formation both by activating necroptosis and by a necroptosis-independent mechanism. Most of the detected ROS originate in mitochondria as a consequence of opening the mitochondrial permeability transition pore. However, a significant part of ROS is produced by mechanisms independent of TNT and necroptosis. Expressing only the tnt gene in Jurkat T-cells also induces lethal ROS formation indicating that these molecular mechanisms are not restricted to macrophages. Both the antioxidant N-acetyl-cysteine and replenishment of NAD+ by providing nicotinamide reduce ROS levels in Mtb-infected macrophages, protect them from cell death, and restrict mycobacterial replication. Our results indicate that a host-directed therapy combining replenishment of NAD+ with inhibition of necroptosis and/or antioxidants might improve the health status of TB patients and augment antibacterial TB chemotherapy.
Keywords: TNT; macrophages; necroptosis; nicotinamide adenine dinucleotide; reactive oxygen species.
© 2019 John Wiley & Sons Ltd.
Conflict of interest statement
Competing financial interests: The authors declare no financial conflict of interest.
Figures
References
-
- Awodele O, Olayemi SO, Nwite JA, & Adeyemo TA (2012). Investigation of the levels of oxidative stress parameters in HIV and HIV-TB co-infected patients. J Infect Dev Ctries, 6(1), 79–85. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
