

Normal Mode Analysis as a Routine Part of a Structural Investigation
- PMID: 31510014
- PMCID: PMC6767145
- DOI: 10.3390/molecules24183293
Normal Mode Analysis as a Routine Part of a Structural Investigation
Abstract
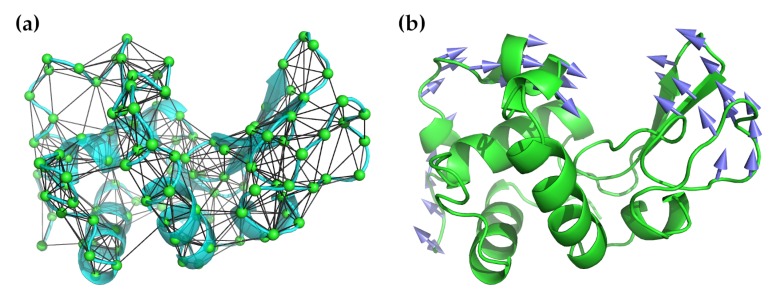
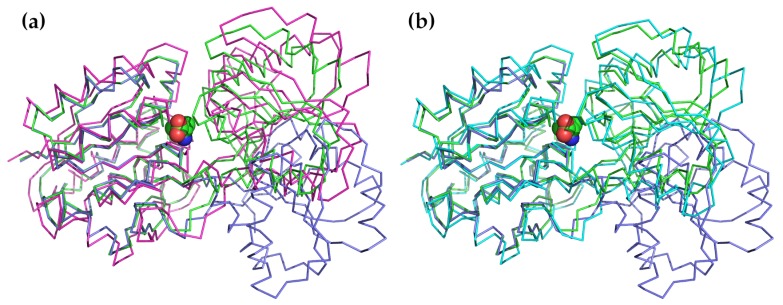
Normal mode analysis (NMA) is a technique that can be used to describe the flexible states accessible to a protein about an equilibrium position. These states have been shown repeatedly to have functional significance. NMA is probably the least computationally expensive method for studying the dynamics of macromolecules, and advances in computer technology and algorithms for calculating normal modes over the last 20 years have made it nearly trivial for all but the largest systems. Despite this, it is still uncommon for NMA to be used as a component of the analysis of a structural study. In this review, we will describe NMA, outline its advantages and limitations, explain what can and cannot be learned from it, and address some criticisms and concerns that have been voiced about it. We will then review the most commonly used techniques for reducing the computational cost of this method and identify the web services making use of these methods. We will illustrate several of their possible uses with recent examples from the literature. We conclude by recommending that NMA become one of the standard tools employed in any structural study.
Keywords: X-ray crystallography; crystal structure; elastic network model; normal mode analysis; protein dynamics.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the preparation, in the writing, or in the decision to publish this manuscript.
Figures
References
-
- Case D.A. Normal mode analysis of protein dynamics. Curr. Opin. Struct. Biol. 1994;4:285–290. doi: 10.1016/S0959-440X(94)90321-2. - DOI
-
- Brooks B.R., Janežič D., Karplus M. Harmonic analysis of large systems. I. Methodology. J. Comput. Chem. 1995;16:1522–1542. doi: 10.1002/jcc.540161209. - DOI
-
- Skjaerven L., Hollup S.M., Reuter N. Normal mode analysis for proteins. J. Mol. Struct. THEOCHEM. 2009;898:42–48. doi: 10.1016/j.theochem.2008.09.024. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
