

Molecular Dynamics Simulation Framework to Probe the Binding Hypothesis of CYP3A4 Inhibitors
- PMID: 31510073
- PMCID: PMC6769491
- DOI: 10.3390/ijms20184468
Molecular Dynamics Simulation Framework to Probe the Binding Hypothesis of CYP3A4 Inhibitors
Abstract
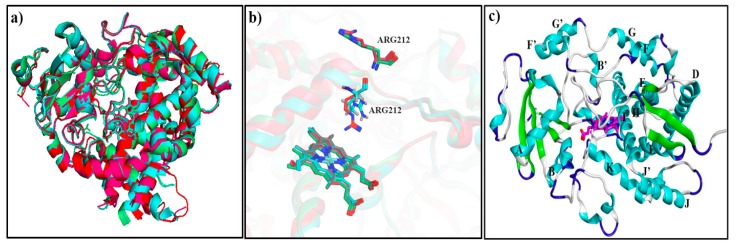
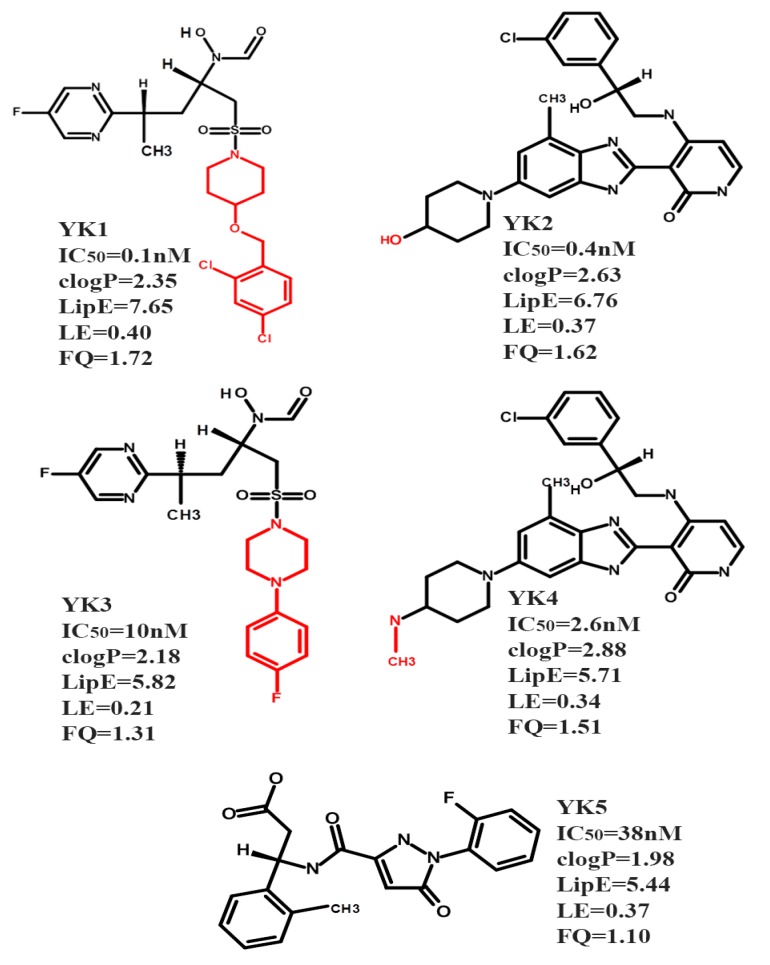
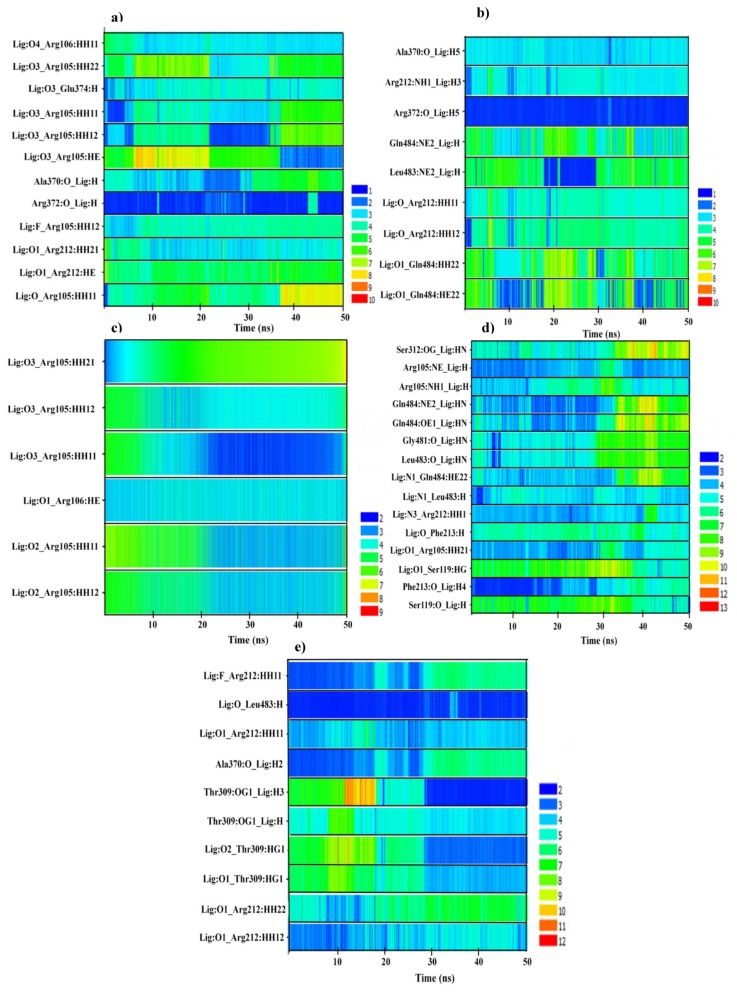
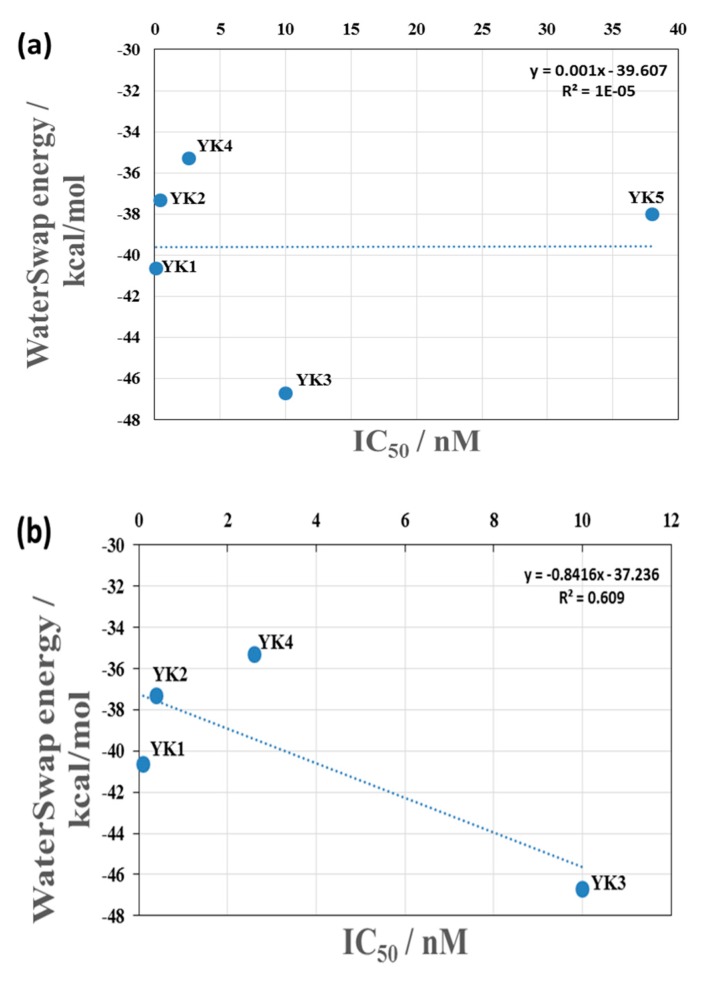
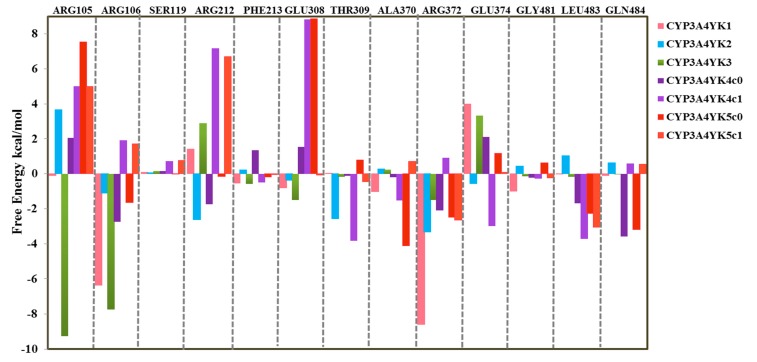
The Cytochrome P450 family of heme-containing proteins plays a major role in catalyzing phase I metabolic reactions, and the CYP3A4 subtype is responsible for the metabolism of many currently marketed drugs. Additionally, CYP3A4 has an inherent affinity for a broad spectrum of structurally diverse chemical entities, often leading to drug-drug interactions mediated by the inhibition or induction of the metabolic enzyme. The current study explores the binding of selected highly efficient CYP3A4 inhibitors by docking and molecular dynamics (MD) simulation protocols and their binding free energy calculated using the WaterSwap method. The results indicate the importance of binding pocket residues including Phe57, Arg105, Arg106, Ser119, Arg212, Phe213, Thr309, Ser312, Ala370, Arg372, Glu374, Gly481 and Leu483 for interaction with CYP3A4 inhibitors. The residue-wise decomposition of the binding free energy from the WaterSwap method revealed the importance of binding site residues Arg106 and Arg372 in the stabilization of all the selected CYP3A4-inhibitor complexes. The WaterSwap binding energies were further complemented with the MM(GB/PB)SA results and it was observed that the binding energies calculated by both methods do not differ significantly. Overall, our results could guide towards the use of multiple computational approaches to achieve a better understanding of CYP3A4 inhibition, subsequently leading to the design of highly specific and efficient new chemical entities with suitable ADMETox properties and reduced side effects.
Keywords: CYP3A4; CYP3A4 inhibitors; WaterSwap; docking; molecular dynamics simulation; residue-wise energy decomposition.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Analysis of CYP3A4-HIV-1 protease drugs interactions by computational methods for Highly Active Antiretroviral Therapy in HIV/AIDS.J Mol Graph Model. 2010 Jan;28(5):455-63. doi: 10.1016/j.jmgm.2009.10.005. Epub 2009 Oct 30. J Mol Graph Model. 2010. PMID: 19931478
-
Computational insights into the different catalytic activities of CYP3A4 and CYP3A5 toward schisantherin E.Chem Biol Drug Des. 2019 May;93(5):854-864. doi: 10.1111/cbdd.13475. Epub 2019 Feb 5. Chem Biol Drug Des. 2019. PMID: 30637977
-
Interaction of Human Drug-Metabolizing CYP3A4 with Small Inhibitory Molecules.Biochemistry. 2019 Feb 19;58(7):930-939. doi: 10.1021/acs.biochem.8b01221. Epub 2019 Jan 24. Biochemistry. 2019. PMID: 30676743 Free PMC article.
-
Ritonavir analogues as a probe for deciphering the cytochrome P450 3A4 inhibitory mechanism.Curr Top Med Chem. 2014;14(11):1348-55. doi: 10.2174/1568026614666140506120647. Curr Top Med Chem. 2014. PMID: 24805065 Free PMC article. Review.
-
Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4.Curr Drug Metab. 2008 May;9(4):310-22. doi: 10.2174/138920008784220664. Curr Drug Metab. 2008. PMID: 18473749 Review.
Cited by
-
Study of tyramine-binding mechanism and insecticidal activity of oil extracted from Eucalyptus against Sitophilus oryzae.Front Chem. 2022 Sep 23;10:964700. doi: 10.3389/fchem.2022.964700. eCollection 2022. Front Chem. 2022. PMID: 36212071 Free PMC article.
-
Single Nucleotide Polymorphism Induces Divergent Dynamic Patterns in CYP3A5: A Microsecond Scale Biomolecular Simulation of Variants Identified in Sub-Saharan African Populations.Int J Mol Sci. 2021 Jul 21;22(15):7786. doi: 10.3390/ijms22157786. Int J Mol Sci. 2021. PMID: 34360551 Free PMC article.
-
A cytochrome P450 insecticide detoxification mechanism is not conserved across the Megachilidae family of bees.Evol Appl. 2023 Dec 6;17(1):e13625. doi: 10.1111/eva.13625. eCollection 2024 Jan. Evol Appl. 2023. PMID: 38283601 Free PMC article.
-
How Plant Polyhydroxy Flavonoids Can Hinder the Metabolism of Cytochrome 3A4.Biomedicines. 2025 Mar 7;13(3):655. doi: 10.3390/biomedicines13030655. Biomedicines. 2025. PMID: 40149631 Free PMC article.
-
Design, synthesis, computational study and cytotoxic evaluation of some new quinazoline derivatives containing pyrimidine moiety.Sci Rep. 2023 Sep 2;13(1):14461. doi: 10.1038/s41598-023-41530-6. Sci Rep. 2023. PMID: 37660139 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
