

Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology
- PMID: 31510660
- PMCID: PMC6612828
- DOI: 10.1093/bioinformatics/btz363
Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology
Abstract
Motivation: The composition and density of immune cells in the tumor microenvironment (TME) profoundly influence tumor progression and success of anti-cancer therapies. Flow cytometry, immunohistochemistry staining or single-cell sequencing are often unavailable such that we rely on computational methods to estimate the immune-cell composition from bulk RNA-sequencing (RNA-seq) data. Various methods have been proposed recently, yet their capabilities and limitations have not been evaluated systematically. A general guideline leading the research community through cell type deconvolution is missing.
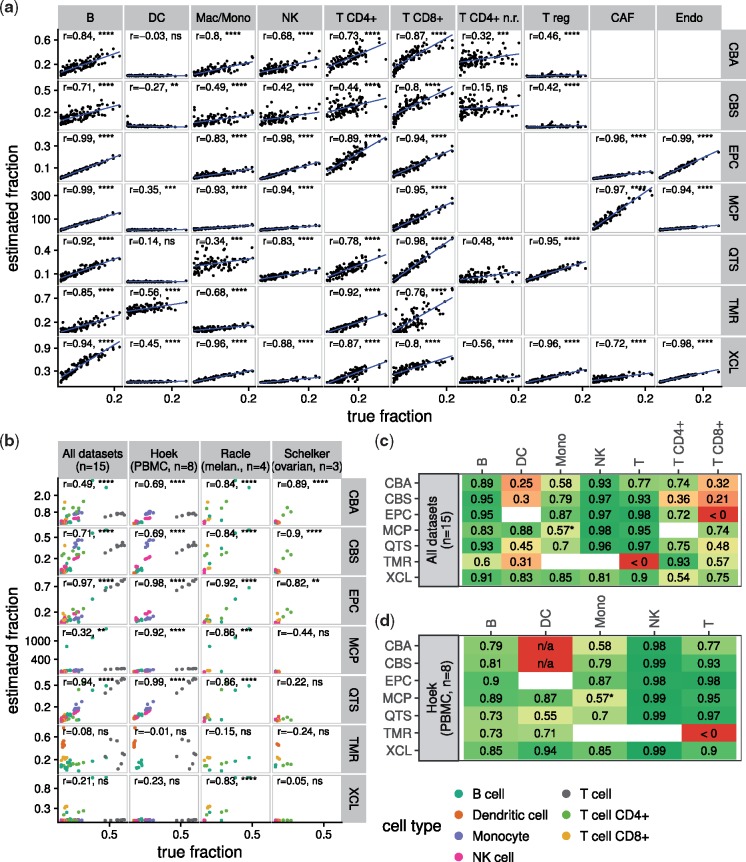
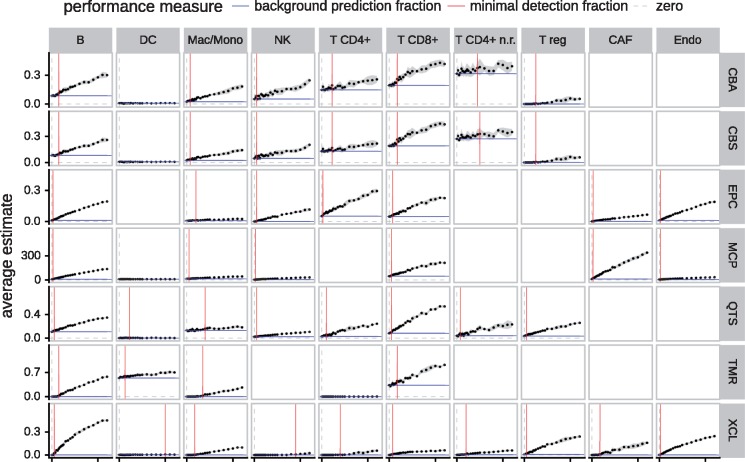
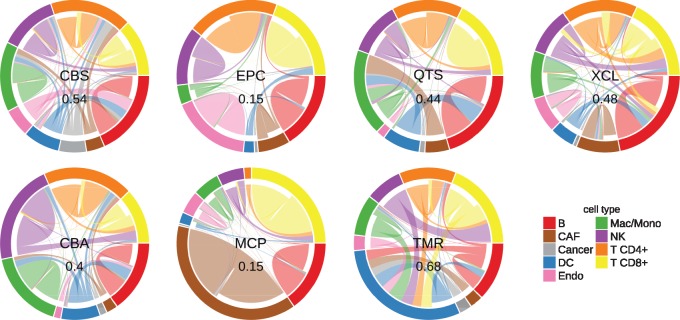
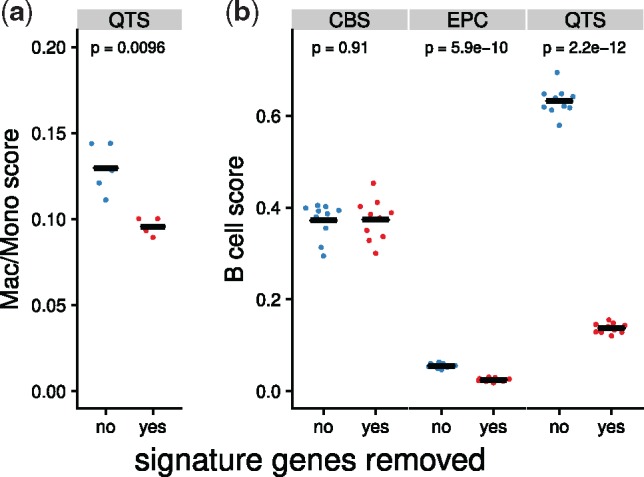
Results: We developed a systematic approach for benchmarking such computational methods and assessed the accuracy of tools at estimating nine different immune- and stromal cells from bulk RNA-seq samples. We used a single-cell RNA-seq dataset of ∼11 000 cells from the TME to simulate bulk samples of known cell type proportions, and validated the results using independent, publicly available gold-standard estimates. This allowed us to analyze and condense the results of more than a hundred thousand predictions to provide an exhaustive evaluation across seven computational methods over nine cell types and ∼1800 samples from five simulated and real-world datasets. We demonstrate that computational deconvolution performs at high accuracy for well-defined cell-type signatures and propose how fuzzy cell-type signatures can be improved. We suggest that future efforts should be dedicated to refining cell population definitions and finding reliable signatures.
Availability and implementation: A snakemake pipeline to reproduce the benchmark is available at https://github.com/grst/immune_deconvolution_benchmark. An R package allows the community to perform integrated deconvolution using different methods (https://grst.github.io/immunedeconv).
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2019. Published by Oxford University Press.
Figures
References
-
- Aran D. (2018) xcell repository on GitHub. https://github.com/dviraran/xCell/blob/ce4d43121c4a161b1e72a50dc875e43d9... (20 September 2018, date last accessed).
-
- Avila Cobos F. et al. (2018) Computational deconvolution of transcriptomics data from mixed cell populations. Bioinformatics, 34, 1969–1979. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
