

Alignment-free filtering for cfNA fusion fragments
- PMID: 31510681
- PMCID: PMC6612805
- DOI: 10.1093/bioinformatics/btz346
Alignment-free filtering for cfNA fusion fragments
Abstract
Motivation: Cell-free nucleic acid (cfNA) sequencing data require improvements to existing fusion detection methods along multiple axes: high depth of sequencing, low allele fractions, short fragment lengths and specialized barcodes, such as unique molecular identifiers.
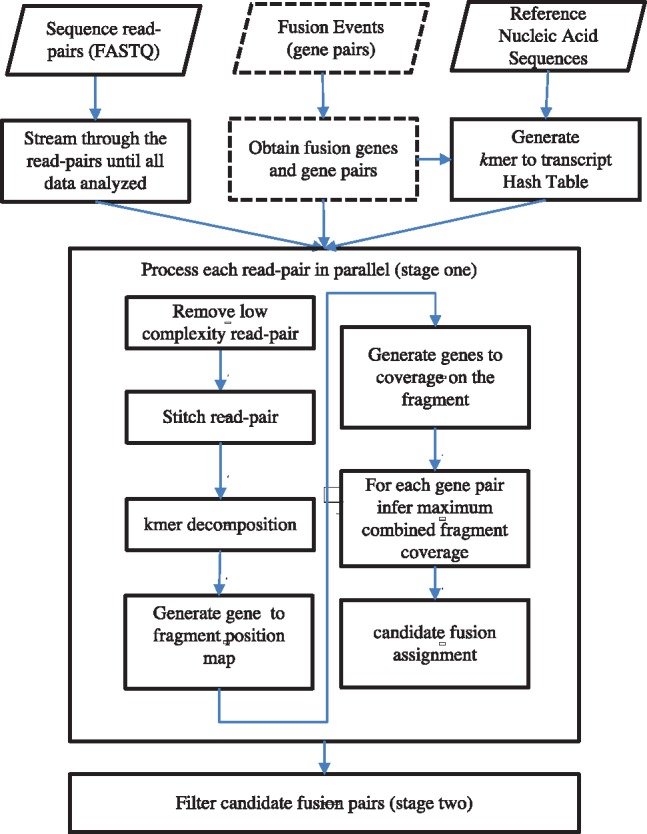
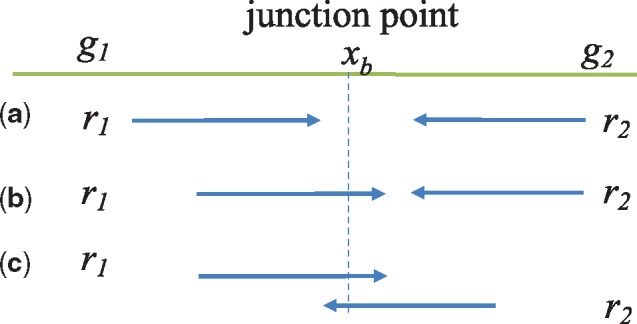
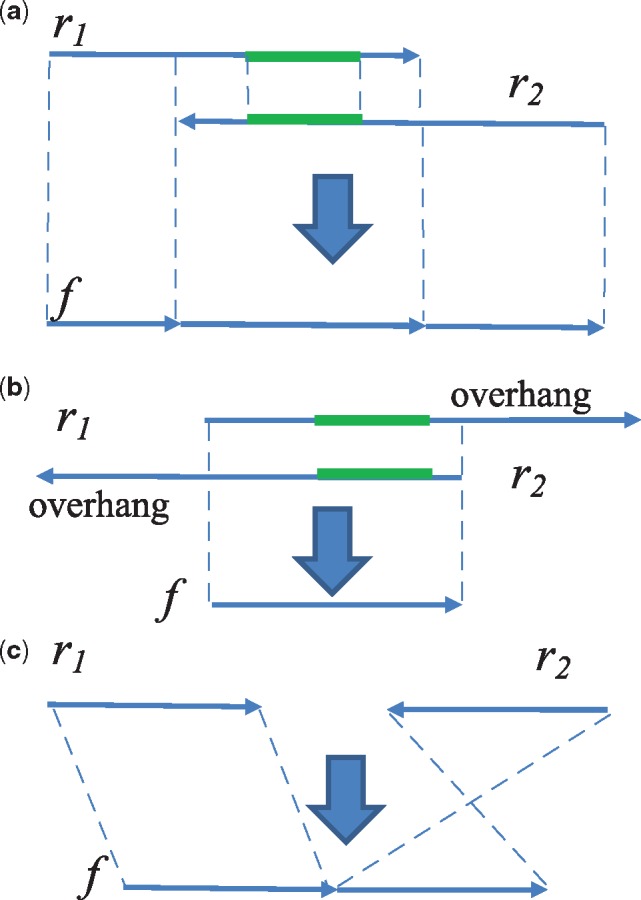
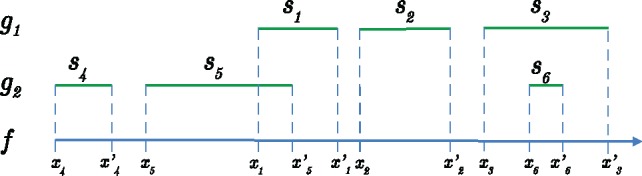
Results: AF4 was developed to address these challenges. It uses a novel alignment-free kmer-based method to detect candidate fusion fragments with high sensitivity and orders of magnitude faster than existing tools. Candidate fragments are then filtered using a max-cover criterion that significantly reduces spurious matches while retaining authentic fusion fragments. This efficient first stage reduces the data sufficiently that commonly used criteria can process the remaining information, or sophisticated filtering policies that may not scale to the raw reads can be used. AF4 provides both targeted and de novo fusion detection modes. We demonstrate both modes in benchmark simulated and real RNA-seq data as well as clinical and cell-line cfNA data.
Availability and implementation: AF4 is open sourced, licensed under Apache License 2.0, and is available at: https://github.com/grailbio/bio/tree/master/fusion.
© The Author(s) 2019. Published by Oxford University Press.
Figures
References
-
- Bray N.L. et al. (2016) Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol., 34, 525.. - PubMed
-
- Donaldson J., Park B.H. (2018) Circulating tumor DNA: measurement and clinical utility. Ann. Rev. Med., 69, 223–234. - PubMed
-
- Haas B. et al. (2017). STAR-Fusion: fast and accurate fusion transcript detection from RNA-seq. https://www.biorxiv.org/content/10.1101/120295v1. - DOI
