

hicGAN infers super resolution Hi-C data with generative adversarial networks
- PMID: 31510693
- PMCID: PMC6612845
- DOI: 10.1093/bioinformatics/btz317
hicGAN infers super resolution Hi-C data with generative adversarial networks
Abstract
Motivation: Hi-C is a genome-wide technology for investigating 3D chromatin conformation by measuring physical contacts between pairs of genomic regions. The resolution of Hi-C data directly impacts the effectiveness and accuracy of downstream analysis such as identifying topologically associating domains (TADs) and meaningful chromatin loops. High resolution Hi-C data are valuable resources which implicate the relationship between 3D genome conformation and function, especially linking distal regulatory elements to their target genes. However, high resolution Hi-C data across various tissues and cell types are not always available due to the high sequencing cost. It is therefore indispensable to develop computational approaches for enhancing the resolution of Hi-C data.
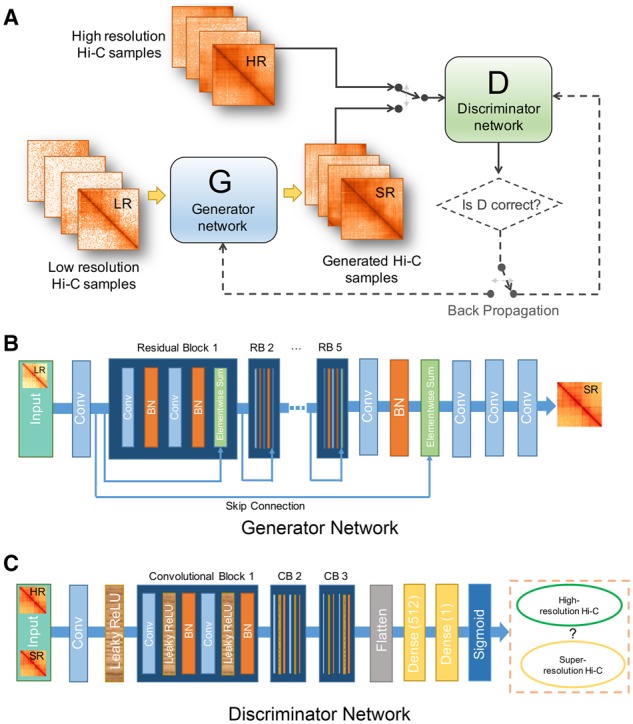
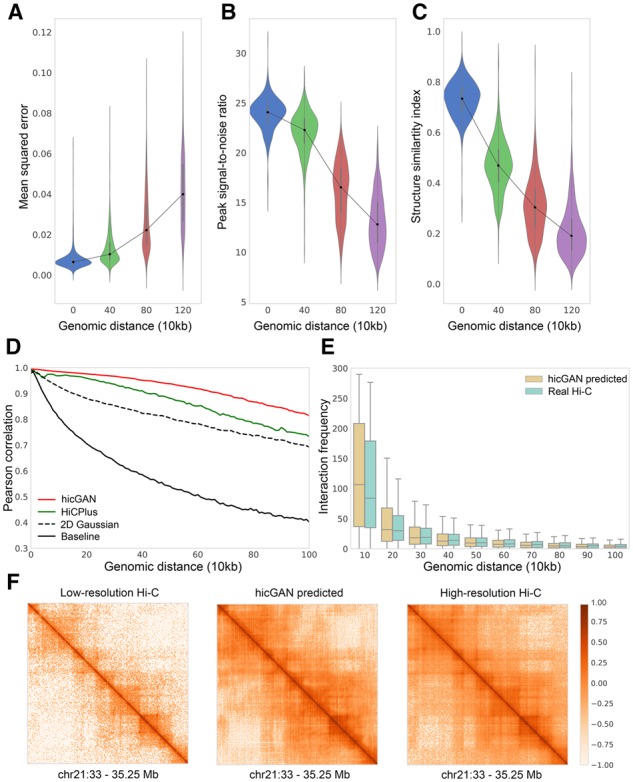
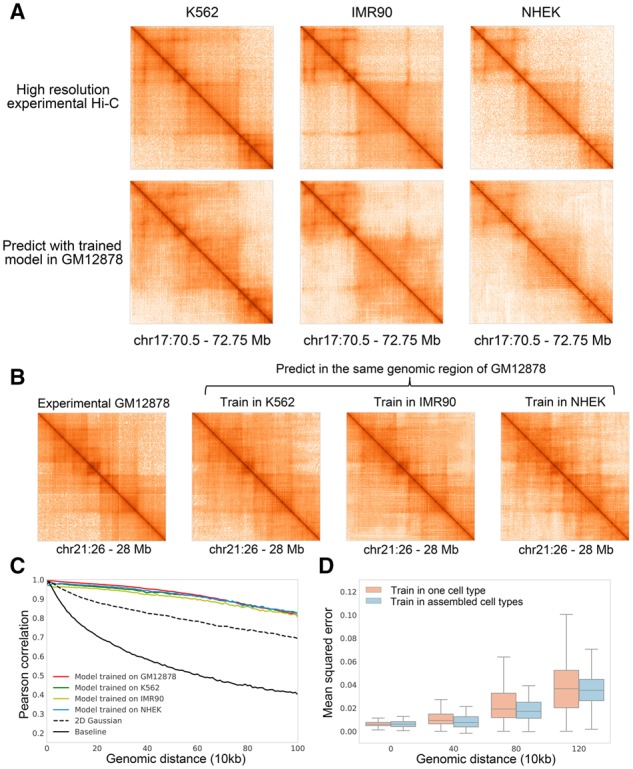
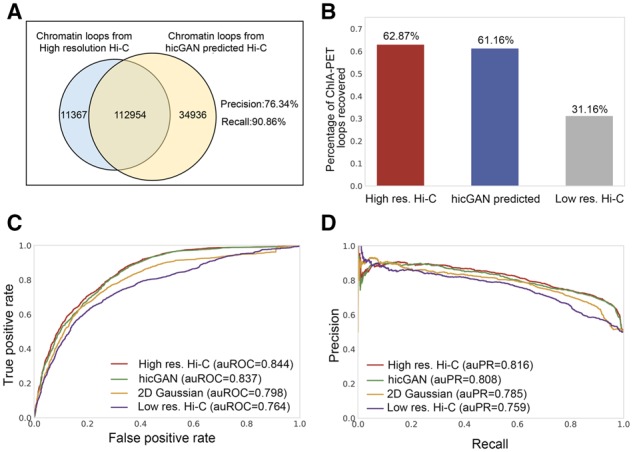
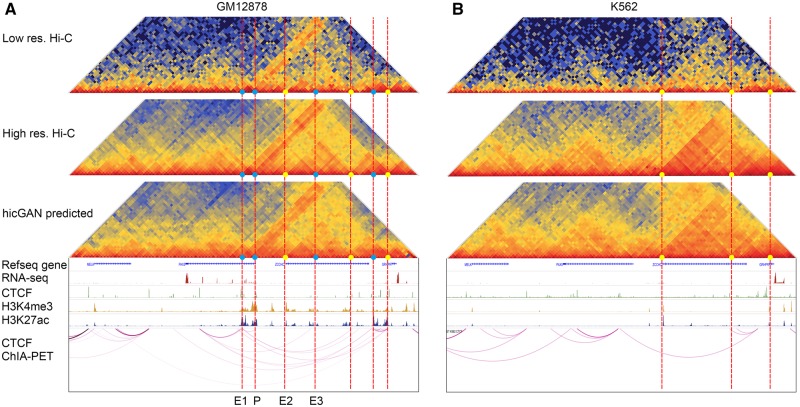
Results: We proposed hicGAN, an open-sourced framework, for inferring high resolution Hi-C data from low resolution Hi-C data with generative adversarial networks (GANs). To the best of our knowledge, this is the first study to apply GANs to 3D genome analysis. We demonstrate that hicGAN effectively enhances the resolution of low resolution Hi-C data by generating matrices that are highly consistent with the original high resolution Hi-C matrices. A typical scenario of usage for our approach is to enhance low resolution Hi-C data in new cell types, especially where the high resolution Hi-C data are not available. Our study not only presents a novel approach for enhancing Hi-C data resolution, but also provides fascinating insights into disclosing complex mechanism underlying the formation of chromatin contacts.
Availability and implementation: We release hicGAN as an open-sourced software at https://github.com/kimmo1019/hicGAN.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2019. Published by Oxford University Press.
Figures
References
-
- Abadi M. et al. (2016) Tensorflow: a system for large-scale machine learning In: OSDI, Savannah, GA, USA, pp. 265–283. USENIX.
-
- Alipanahi B. et al. (2015) Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol., 33, 831–838. - PubMed
-
- Dekker J. et al. (2002) Capturing chromosome conformation. Science, 295, 1306–1311. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
