

Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials
- PMID: 31511426
- PMCID: PMC7717492
- DOI: 10.1126/scitranslmed.aaw8412
Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials
Abstract
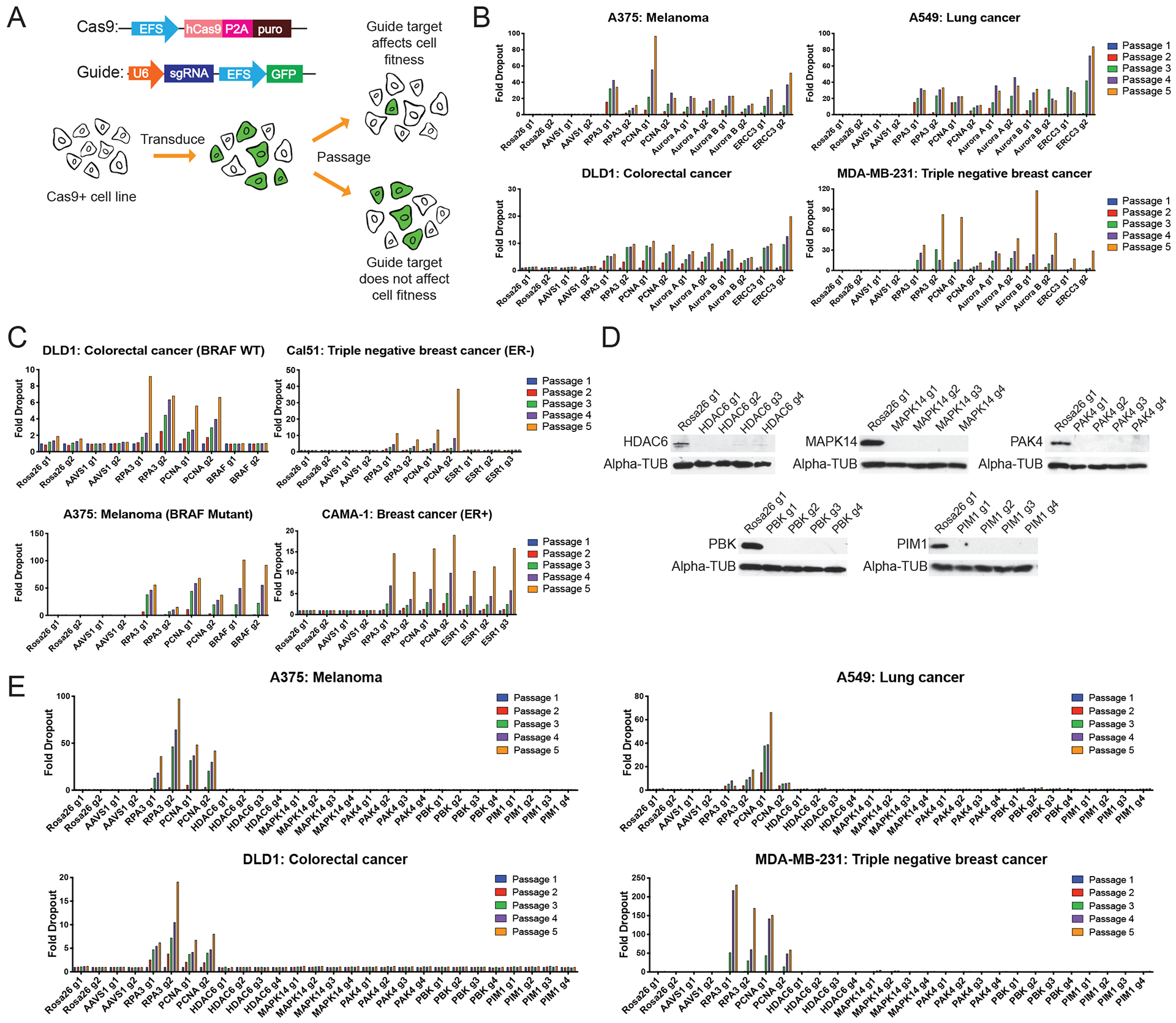
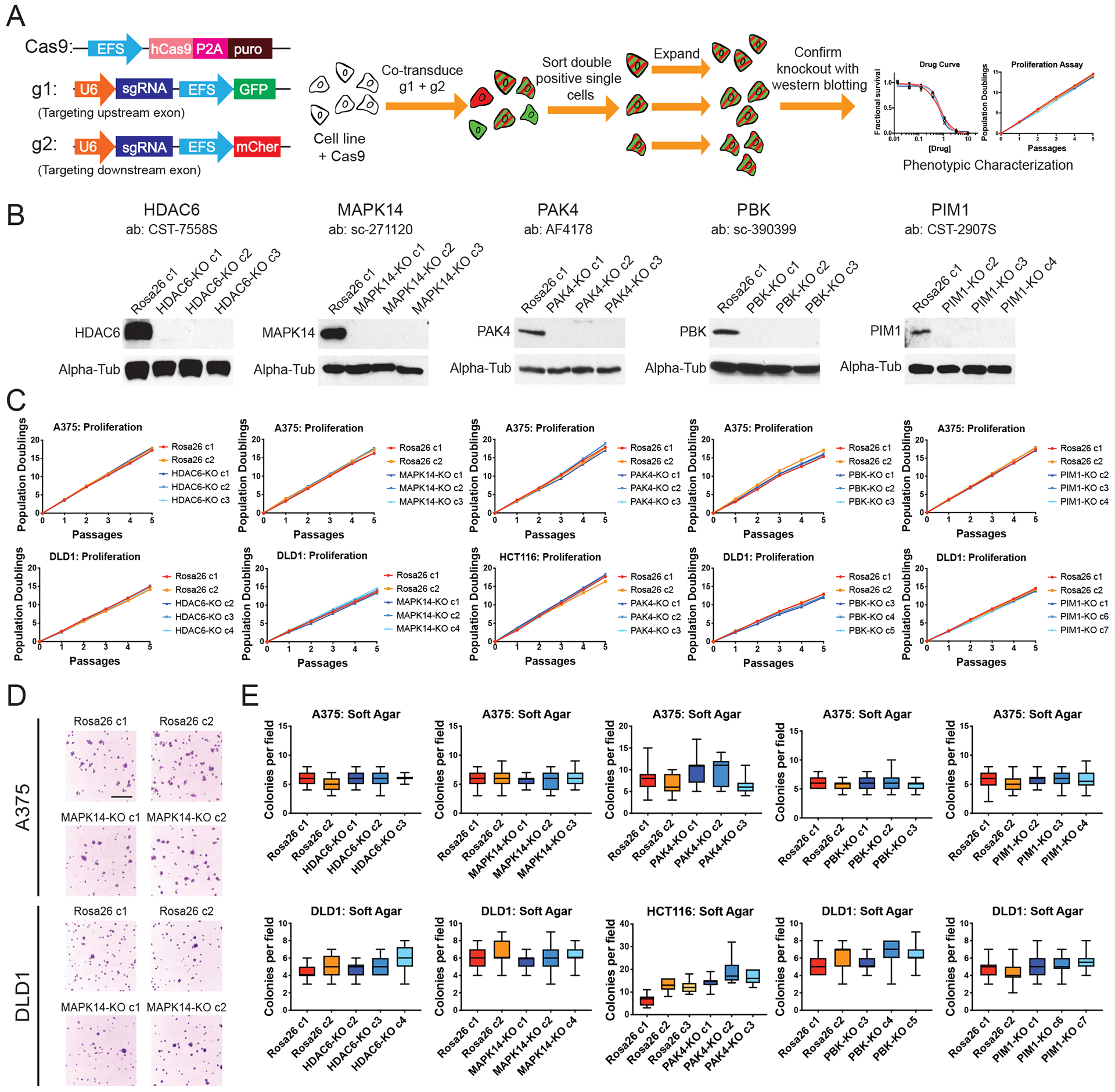
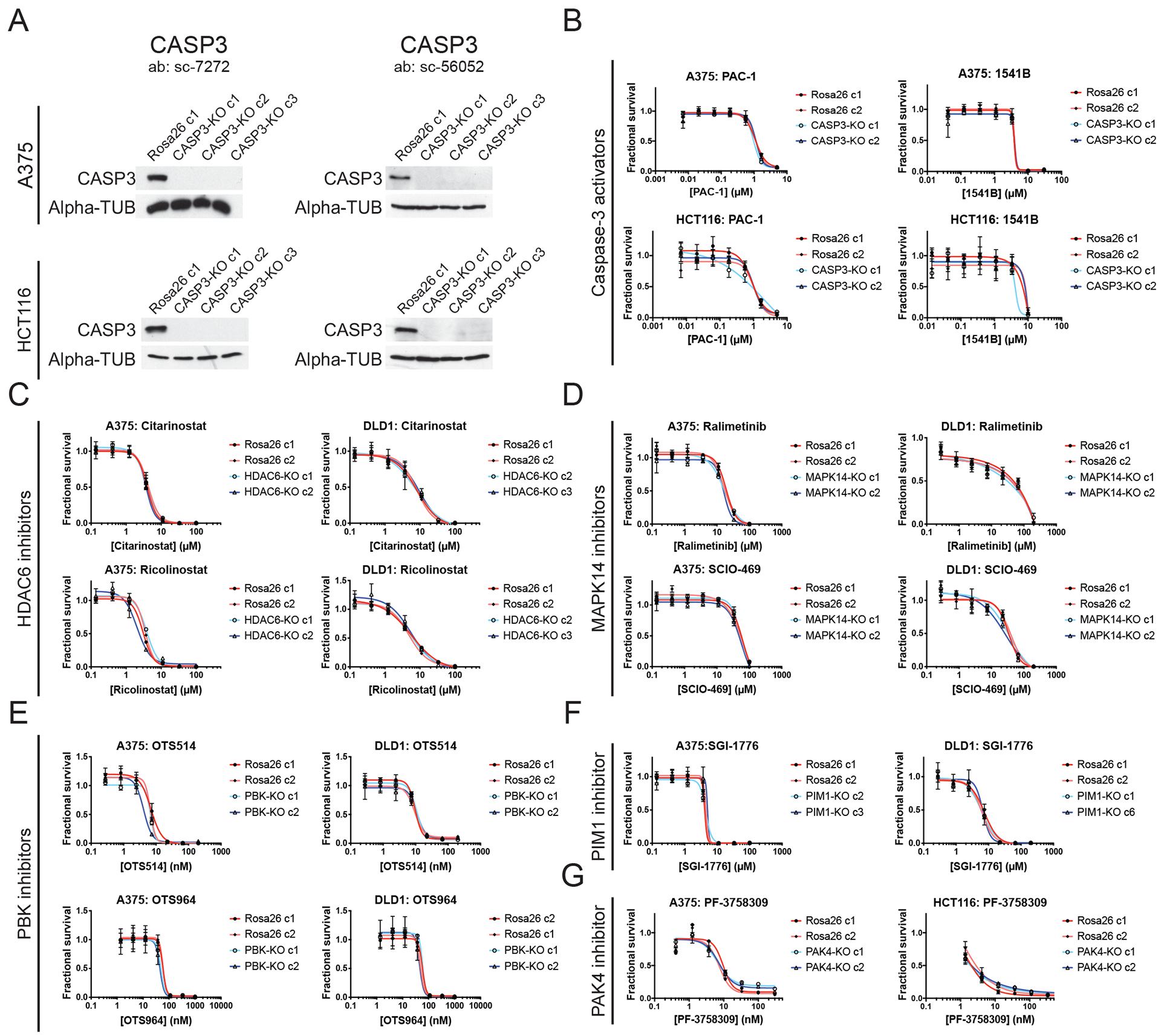
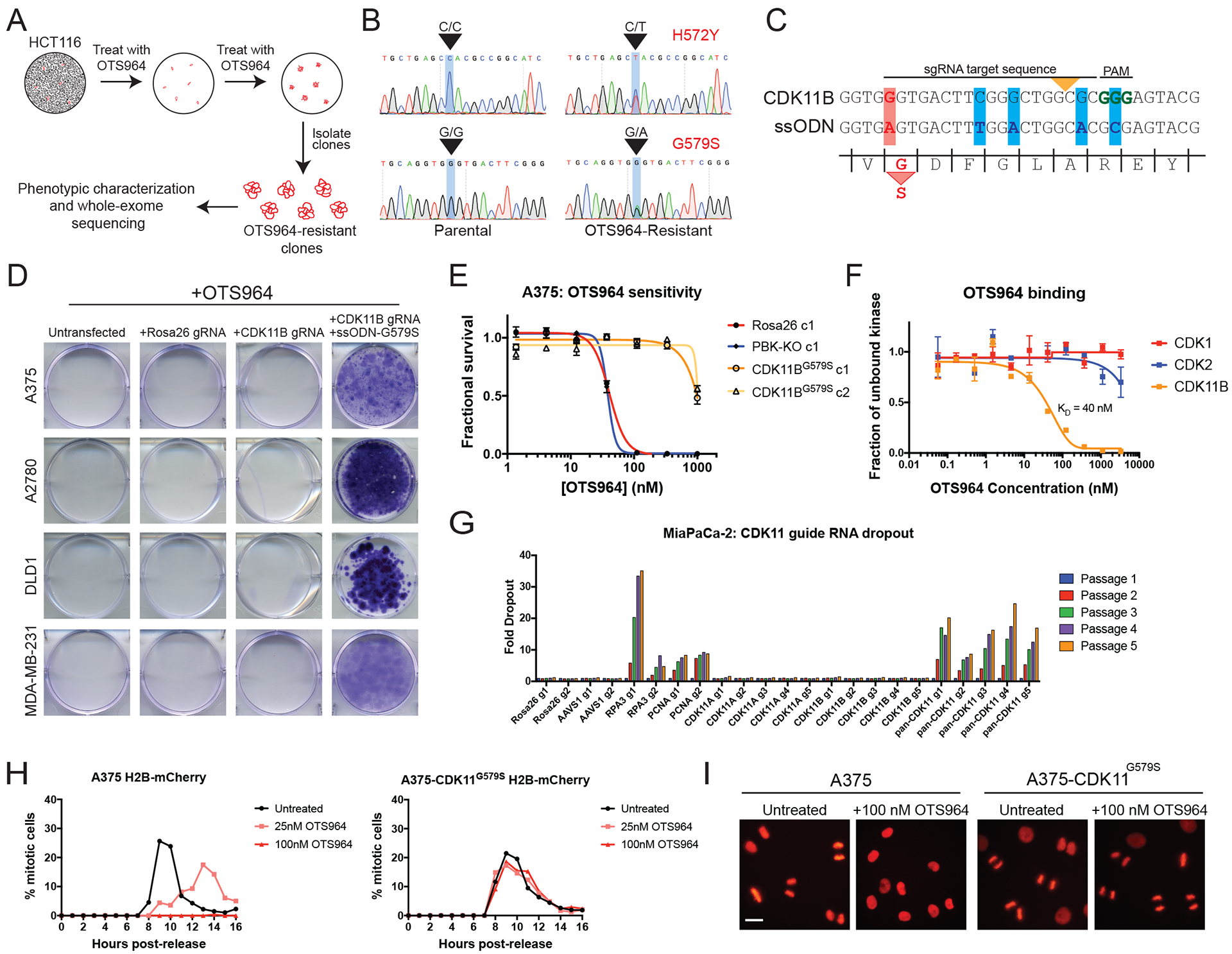
Ninety-seven percent of drug-indication pairs that are tested in clinical trials in oncology never advance to receive U.S. Food and Drug Administration approval. While lack of efficacy and dose-limiting toxicities are the most common causes of trial failure, the reason(s) why so many new drugs encounter these problems is not well understood. Using CRISPR-Cas9 mutagenesis, we investigated a set of cancer drugs and drug targets in various stages of clinical testing. We show that-contrary to previous reports obtained predominantly with RNA interference and small-molecule inhibitors-the proteins ostensibly targeted by these drugs are nonessential for cancer cell proliferation. Moreover, the efficacy of each drug that we tested was unaffected by the loss of its putative target, indicating that these compounds kill cells via off-target effects. By applying a genetic target-deconvolution strategy, we found that the mischaracterized anticancer agent OTS964 is actually a potent inhibitor of the cyclin-dependent kinase CDK11 and that multiple cancer types are addicted to CDK11 expression. We suggest that stringent genetic validation of the mechanism of action of cancer drugs in the preclinical setting may decrease the number of therapies tested in human patients that fail to provide any clinical benefit.
Copyright © 2019 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
Figures
Comment in
-
Avoiding target misidentification.Nat Rev Drug Discov. 2019 Oct;18(11):826. doi: 10.1038/d41573-019-00161-1. Nat Rev Drug Discov. 2019. PMID: 31673126 No abstract available.
References
-
- Sharma SV, Settleman J, Oncogene addiction: setting the stage for molecularly targeted cancer therapy, Genes Dev. 21, 3214–3231 (2007). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
