

The endotoxin hypothesis of neurodegeneration
- PMID: 31519175
- PMCID: PMC6744684
- DOI: 10.1186/s12974-019-1564-7
The endotoxin hypothesis of neurodegeneration
Abstract
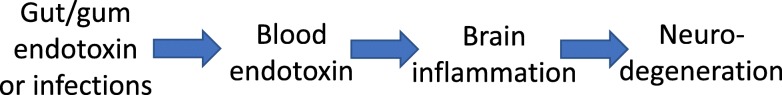
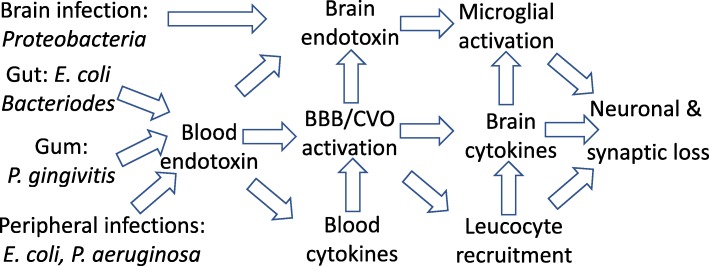
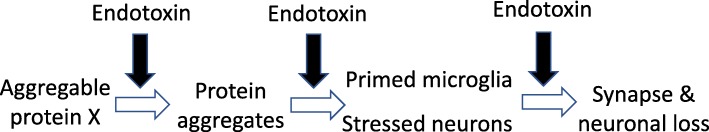
The endotoxin hypothesis of neurodegeneration is the hypothesis that endotoxin causes or contributes to neurodegeneration. Endotoxin is a lipopolysaccharide (LPS), constituting much of the outer membrane of gram-negative bacteria, present at high concentrations in gut, gums and skin and in other tissue during bacterial infection. Blood plasma levels of endotoxin are normally low, but are elevated during infections, gut inflammation, gum disease and neurodegenerative disease. Adding endotoxin at such levels to blood of healthy humans induces systemic inflammation and brain microglial activation. Adding high levels of endotoxin to the blood or body of rodents induces microglial activation, priming and/or tolerance, memory deficits and loss of brain synapses and neurons. Endotoxin promotes amyloid β and tau aggregation and neuropathology, suggesting the possibility that endotoxin synergises with different aggregable proteins to give different neurodegenerative diseases. Blood and brain endotoxin levels are elevated in Alzheimer's disease, which is accelerated by systemic infections, including gum disease. Endotoxin binds directly to APOE, and the APOE4 variant both sensitises to endotoxin and predisposes to Alzheimer's disease. Intestinal permeability increases early in Parkinson's disease, and injection of endotoxin into mice induces α-synuclein production and aggregation, as well as loss of dopaminergic neurons in the substantia nigra. The gut microbiome changes in Parkinson's disease, and changing the endotoxin-producing bacterial species can affect the disease in patients and mouse models. Blood endotoxin is elevated in amyotrophic lateral sclerosis, and endotoxin promotes TDP-43 aggregation and neuropathology. Peripheral diseases that elevate blood endotoxin, such as sepsis, AIDS and liver failure, also result in neurodegeneration. Endotoxin directly and indirectly activates microglia that damage neurons via nitric oxide, oxidants and cytokines, and by phagocytosis of synapses and neurons. The endotoxin hypothesis is unproven, but if correct, then neurodegeneration may be reduced by decreasing endotoxin levels or endotoxin-induced neuroinflammation.
Keywords: Alzheimer’s disease; Bacteria; Endotoxin; Gut microbiome; Inflammation; Lipopolysaccharide; Microglia; Neurodegeneration; Neuroinflammation; Parkinson’s disease.
Conflict of interest statement
The author declares that he/she has no competing interests.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
