

A small gene sequencing panel realises a high diagnostic rate in patients with congenital nystagmus following basic phenotyping
- PMID: 31519934
- PMCID: PMC6744446
- DOI: 10.1038/s41598-019-49368-7
A small gene sequencing panel realises a high diagnostic rate in patients with congenital nystagmus following basic phenotyping
Abstract
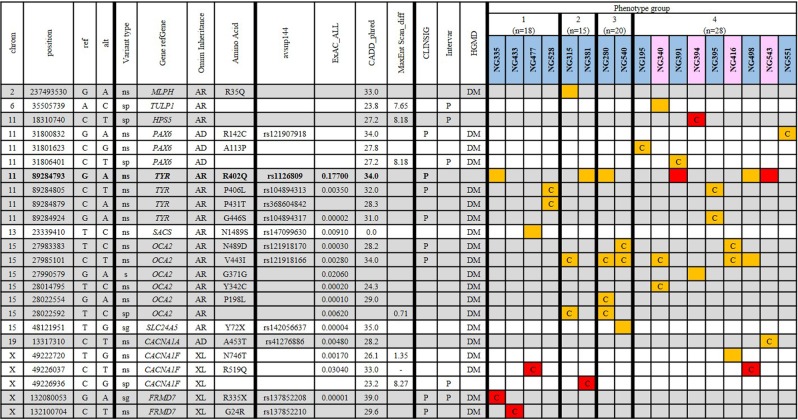
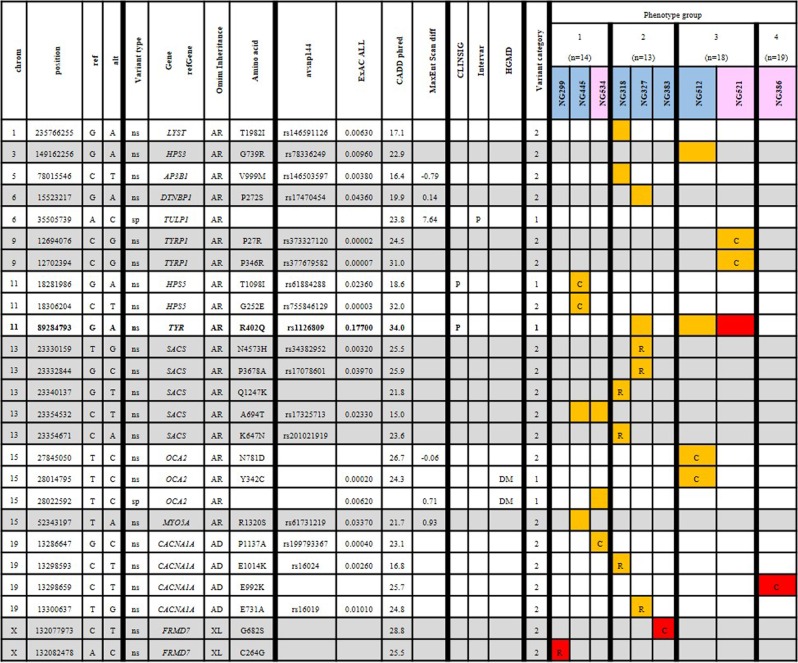
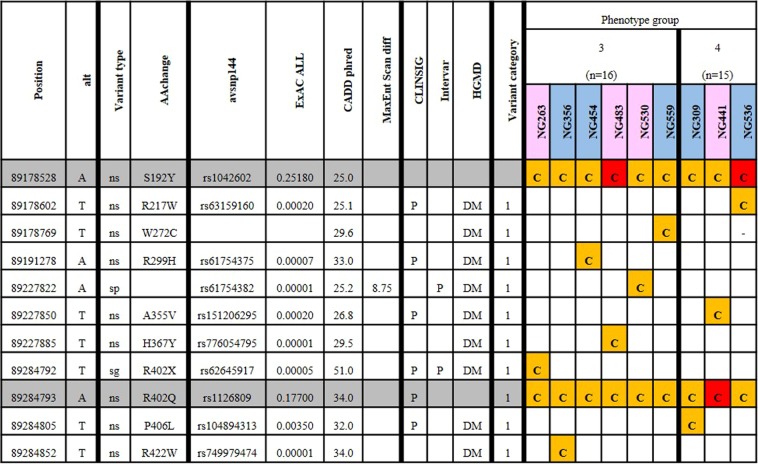
Nystagmus is a disorder of uncontrolled eye movement and can occur as an isolated trait (idiopathic INS, IINS) or as part of multisystem disorders such as albinism, significant visual disorders or neurological disease. Eighty-one unrelated patients with nystagmus underwent routine ocular phenotyping using commonly available phenotyping methods and were grouped into four sub-cohorts according to the level of phenotyping information gained and their findings. DNA was extracted and sequenced using a broad utility next generation sequencing (NGS) gene panel. A clinical subpanel of genes for nystagmus/albinism was utilised and likely causal variants were prioritised according to methods currently employed by clinical diagnostic laboratories. We determine the likely underlying genetic cause for 43.2% of participants with similar yields regardless of prior phenotyping. This study demonstrates that a diagnostic workflow combining basic ocular phenotyping and a clinically available targeted NGS panel, can provide a high diagnostic yield for patients with infantile nystagmus, enabling access to disease specific management at a young age and reducing the need for multiple costly, often invasive tests. By describing diagnostic yield for groups of patients with incomplete phenotyping data, it also permits the subsequent design of 'real-world' diagnostic workflows and illustrates the changing role of genetic testing in modern diagnostic workflows for heterogeneous ophthalmic disorders.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- CEMAS Workshop. Classification of Eye Movement Abnormalities and Strabismus (CEMAS) Workshop report (2001).
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
