

High-Throughput Assessment of Kinome-wide Activation States
- PMID: 31521607
- PMCID: PMC6838672
- DOI: 10.1016/j.cels.2019.08.005
High-Throughput Assessment of Kinome-wide Activation States
Abstract
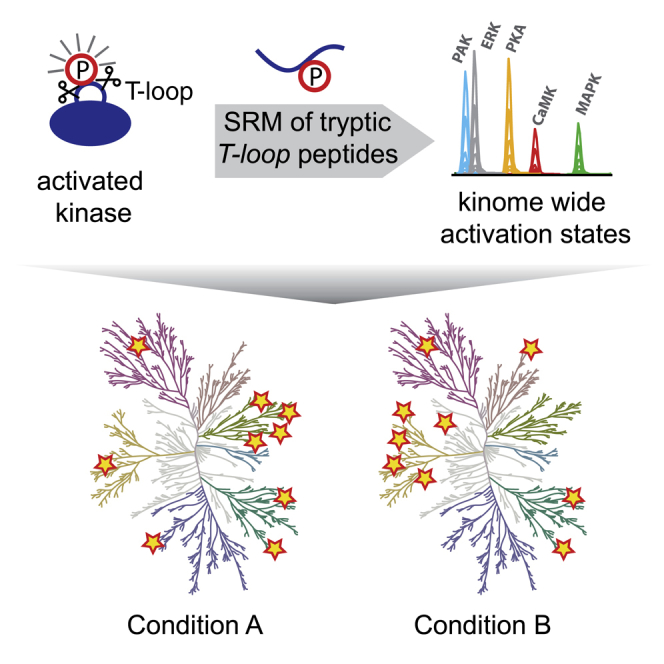
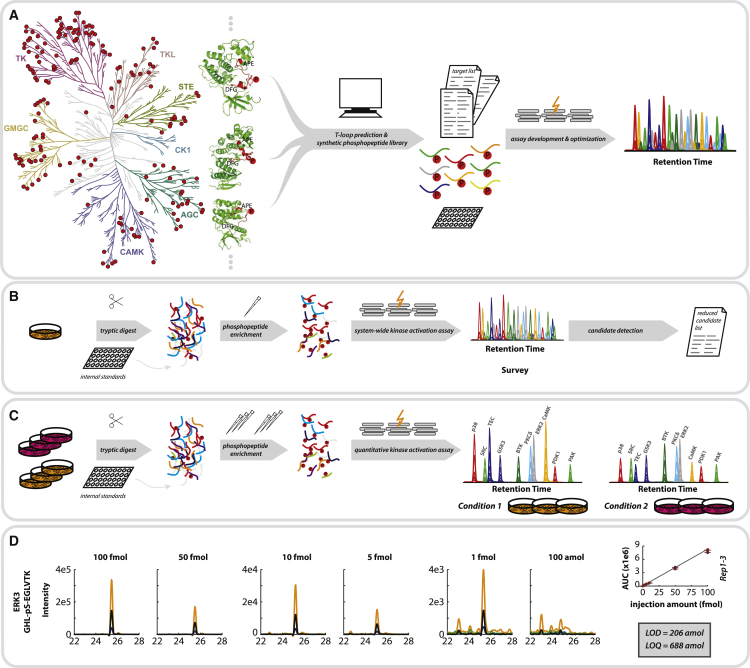
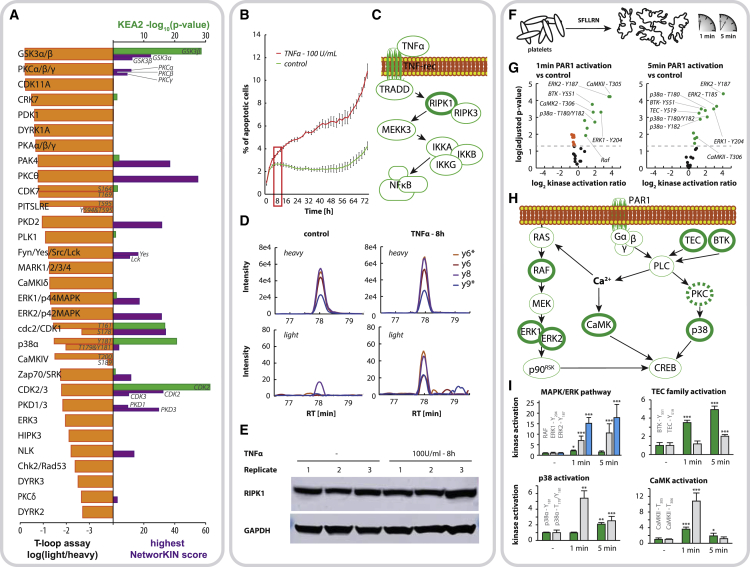
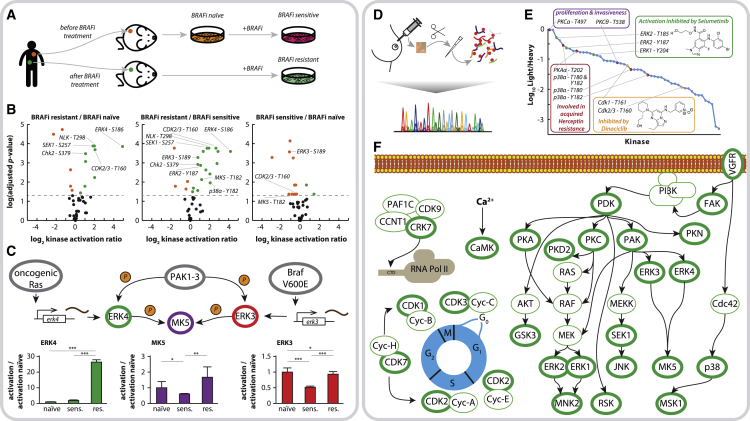
Aberrant kinase activity has been linked to a variety of disorders; however, methods to probe kinase activation states in cells have been lacking. Until now, kinase activity has mainly been deduced from either protein expression or substrate phosphorylation levels. Here, we describe a strategy to directly infer kinase activation through targeted quantification of T-loop phosphorylation, which serves as a critical activation switch in a majority of protein kinases. Combining selective phosphopeptide enrichment with robust targeted mass spectrometry, we provide highly specific assays for 248 peptides, covering 221 phosphosites in the T-loop region of 178 human kinases. Using these assays, we monitored the activation of 63 kinases through 73 T-loop phosphosites across different cell types, primary cells, and patient-derived tissue material. The sensitivity of our assays is highlighted by the reproducible detection of TNF-α-induced RIPK1 activation and the detection of 46 T-loop phosphorylation sites from a breast tumor needle biopsy.
Keywords: SRM; T-loop phosphorylation; cancer; kinase; kinase activity; phosphoproteomics; proteomics; signaling; targeted mass spectrometry.
Copyright © 2019 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Patent applicant, Utrecht Holding; name of inventor(s), M.A. and T.S.; application number, EP18179104.7; status of application, pending; specific aspect of the manuscript covered in the patent application, method for monitoring kinase activity in a sample. M.A. is a scientific advisor for Pepscope.
Figures
References
-
- Abelin J.G., Patel J., Lu X., Feeney C.M., Fagbami L., Creech A.L., Hu R., Lam D., Davison D., Pino L. Reduced-representation phosphosignatures measured by quantitative targeted ms capture cellular states and enable large-scale comparison of drug-induced phenotypes. Mol. Cell. Proteomics. 2016;15:1622–1641. - PMC - PubMed
-
- Annibaldi A., Meier P. Checkpoints in TNF-induced cell death: implications in inflammation and cancer. Trends Mol. Med. 2018;24:49–65. - PubMed
-
- Atkinson B.T., Ellmeier W., Watson S.P. Tec regulates platelet activation by GPVI in the absence of Btk. Blood. 2003;102:3592–3599. - PubMed
-
- Bantscheff M., Eberhard D., Abraham Y., Bastuck S., Boesche M., Hobson S., Mathieson T., Perrin J., Raida M., Rau C. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol. 2007;25:1035–1044. - PubMed
-
- Batth T.S., Francavilla C., Olsen J.V. Off-line high-pH reversed-phase fractionation for in-depth phosphoproteomics. J. Proteome Res. 2014;13:6176–6186. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
