

Progress in the understanding and treatment of Fabry disease
- PMID: 31526868
- PMCID: PMC6981246
- DOI: 10.1016/j.bbagen.2019.129437
Progress in the understanding and treatment of Fabry disease
Abstract
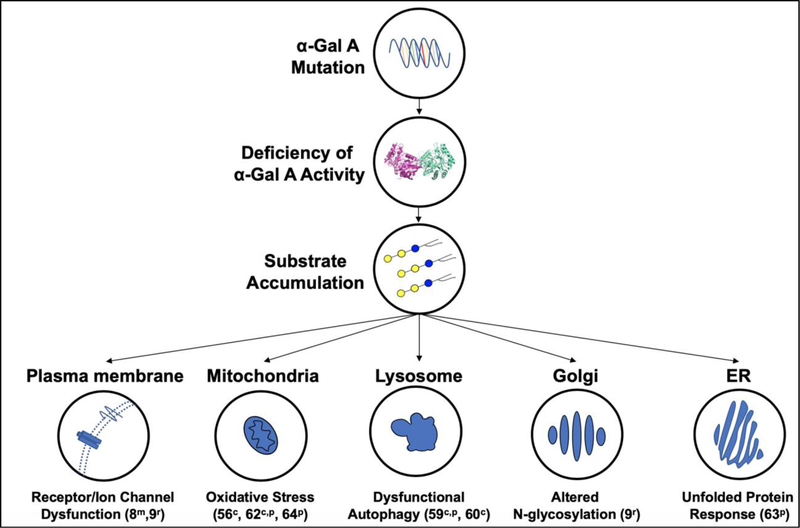
Background: Fabry disease is caused by α-galactosidase A deficiency. Substrates of this lysosomal enzyme accumulate, resulting in cellular dysfunction. Patients experience neuropathic pain, kidney failure, heart disease, and strokes.
Scope of review: The clinical picture and molecular features of Fabry disease are described, along with updates on disease mechanisms, animal models, and therapies.
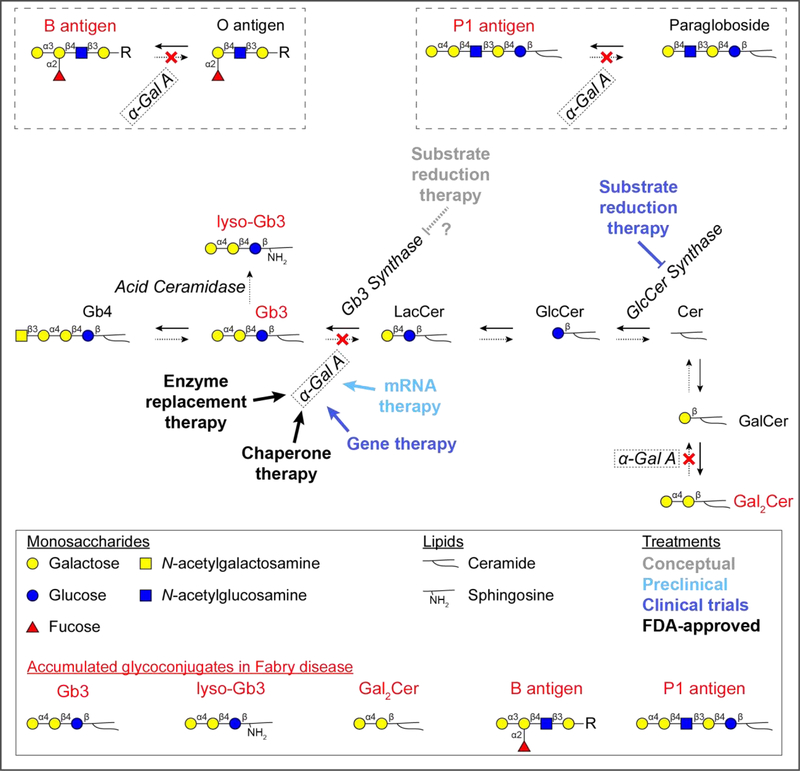
Major conclusions: How the accumulation of α-galactosidase A substrates, mainly glycosphingolipids, leads to organ damage is incompletely understood. Enzyme replacement and chaperone therapies are clinically available to patients, while substrate reduction, mRNA-based, and gene therapies are on the horizon. Animal models exist to optimize these therapies and elucidate disease mechanisms for novel treatments.
General significance: Recent newborn screening studies demonstrate that Fabry disease is the most common lysosomal storage disease. As many countries now include Fabry disease in their screening panels, the number of identified patients is expected to increase significantly. Better knowledge of disease pathogenesis is needed to improve treatment options.
Keywords: Chaperone therapy; Enzyme replacement therapy; Fabry disease; Glycosphingolipids; Lysosomal storage disease; Rodent models.
Copyright © 2019 Elsevier B.V. All rights reserved.
Figures
References
-
- Anderson W. A case of “angio-keratoma”. Br J Dermatol. 1898;10(4):113–7. doi: doi:10.1111/j.1365-2133.1898.tb16317.x. - DOI
-
- Fabry J. Ein Beitrag zur Kenntnis der Purpura haemorrhagica nodularis (Purpura papulosa haemorrhagica Hebrae). Archiv für Dermatologie und Syphilis, Berlin. 1898;43:187–200.
-
- Sweeley CC, Klionsky B. Fabry’s Disease: Classification as a Sphingolipidosis and Partial Characterization of a Novel Glycolipid. J Biol Chem. 1963;238:3148–50. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
