

cellHarmony: cell-level matching and holistic comparison of single-cell transcriptomes
- PMID: 31529053
- PMCID: PMC6868361
- DOI: 10.1093/nar/gkz789
cellHarmony: cell-level matching and holistic comparison of single-cell transcriptomes
Abstract
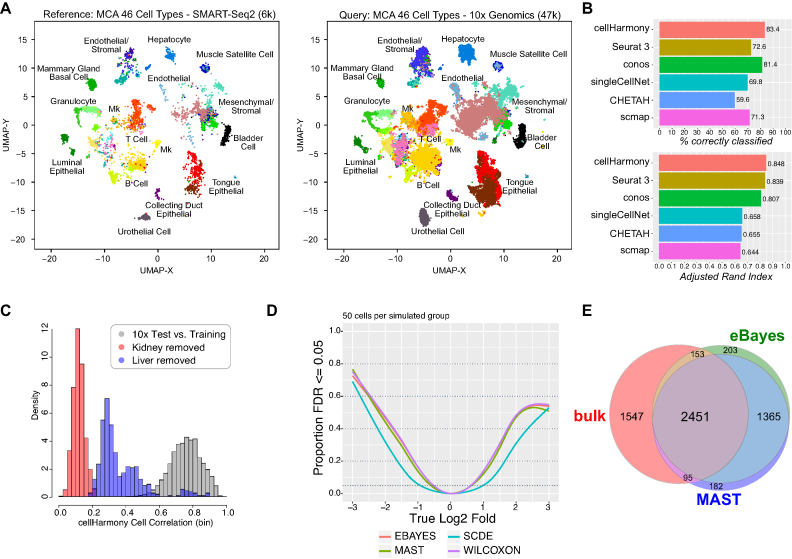
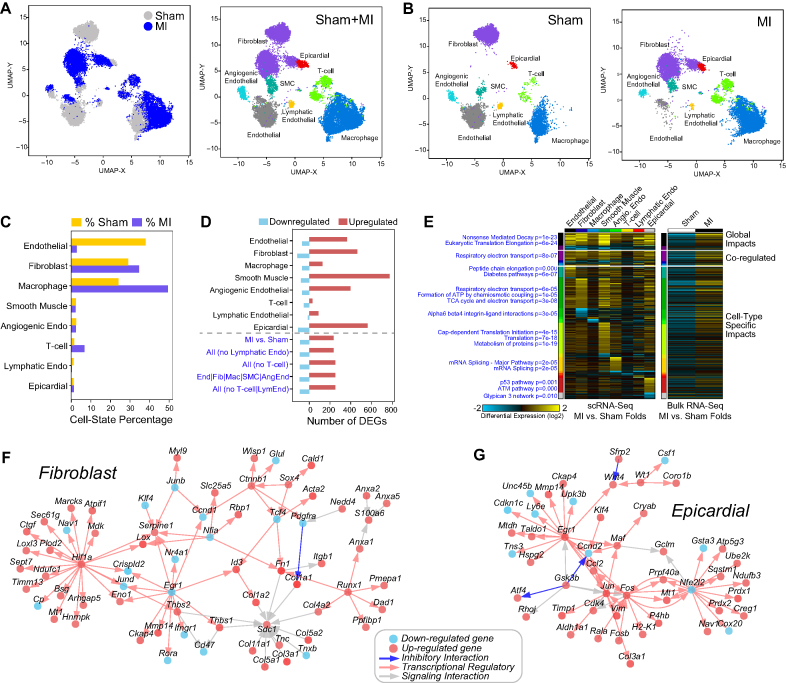
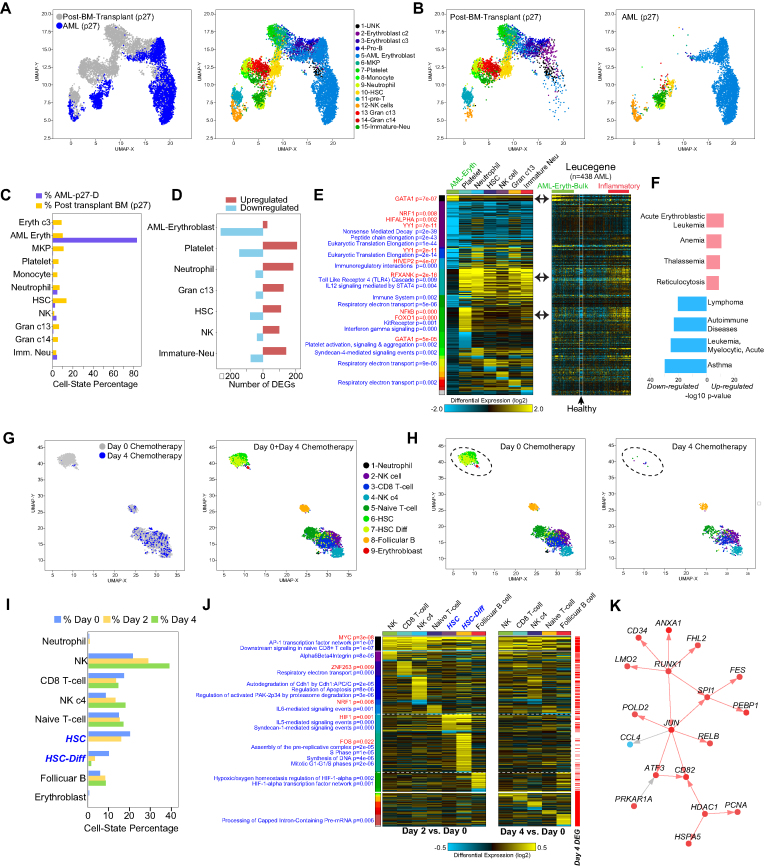
To understand the molecular pathogenesis of human disease, precision analyses to define alterations within and between disease-associated cell populations are desperately needed. Single-cell genomics represents an ideal platform to enable the identification and comparison of normal and diseased transcriptional cell populations. We created cellHarmony, an integrated solution for the unsupervised analysis, classification, and comparison of cell types from diverse single-cell RNA-Seq datasets. cellHarmony efficiently and accurately matches single-cell transcriptomes using a community-clustering and alignment strategy to compute differences in cell-type specific gene expression over potentially dozens of cell populations. Such transcriptional differences are used to automatically identify distinct and shared gene programs among cell-types and identify impacted pathways and transcriptional regulatory networks to understand the impact of perturbations at a systems level. cellHarmony is implemented as a python package and as an integrated workflow within the software AltAnalyze. We demonstrate that cellHarmony has improved or equivalent performance to alternative label projection methods, is able to identify the likely cellular origins of malignant states, stratify patients into clinical disease subtypes from identified gene programs, resolve discrete disease networks impacting specific cell-types, and illuminate therapeutic mechanisms. Thus, this approach holds tremendous promise in revealing the molecular and cellular origins of complex disease.
© The Author(s) 2019. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures


References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
