

Therapeutic Modulation of the Immune Response in Arrhythmogenic Cardiomyopathy
- PMID: 31533459
- PMCID: PMC6817418
- DOI: 10.1161/CIRCULATIONAHA.119.040676
Therapeutic Modulation of the Immune Response in Arrhythmogenic Cardiomyopathy
Abstract
Background: Inflammation is a prominent feature of arrhythmogenic cardiomyopathy (ACM), but whether it contributes to the disease phenotype is not known.
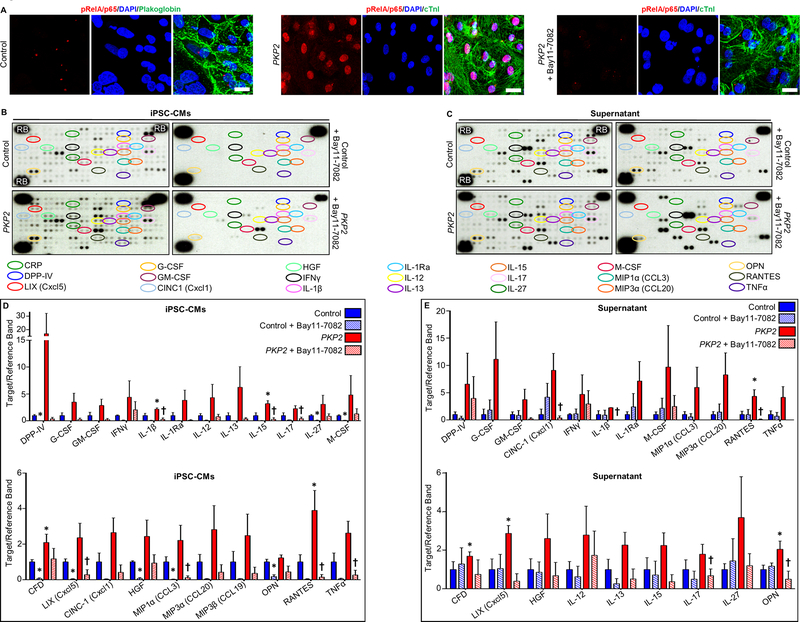
Methods: To define the role of inflammation in the pathogenesis of ACM, we characterized nuclear factor-κB signaling in ACM models in vitro and in vivo and in cardiac myocytes from patient induced pluripotent stem cells.
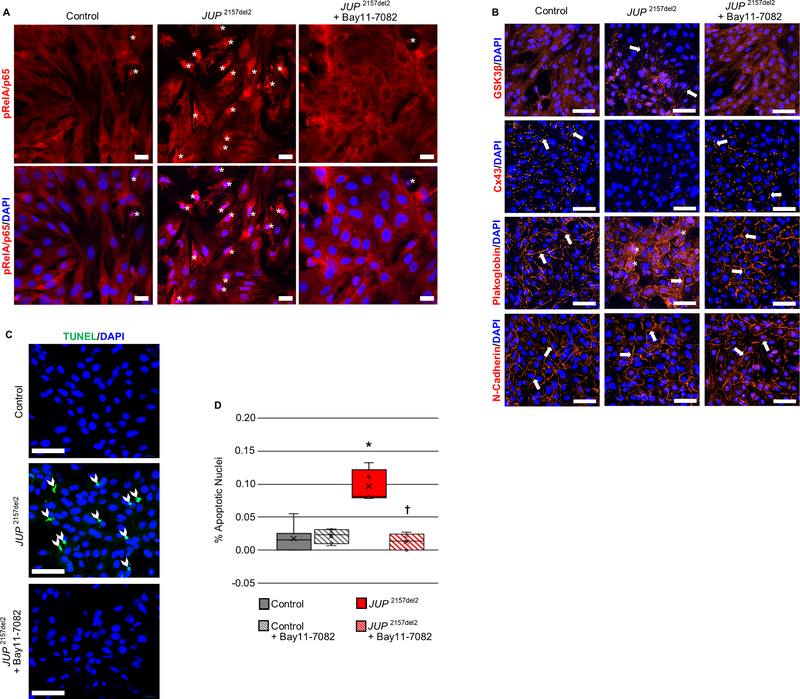
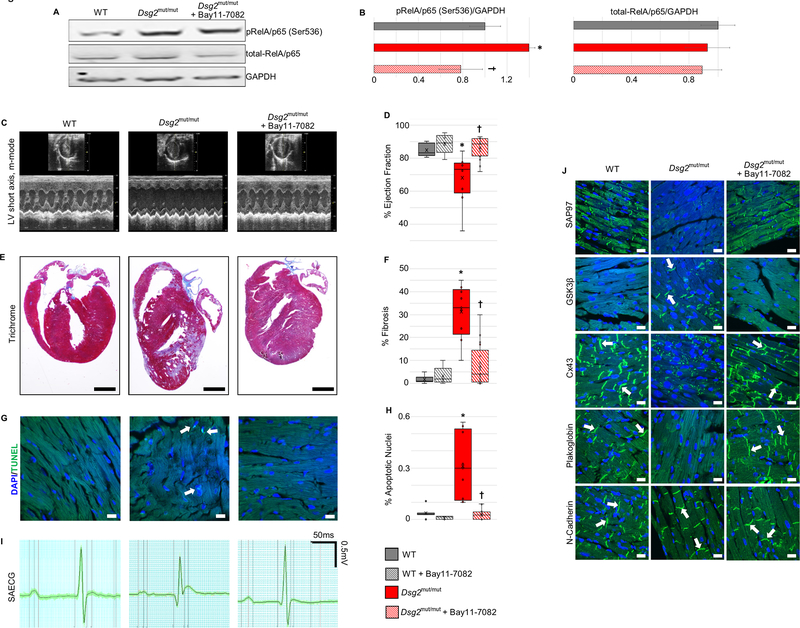
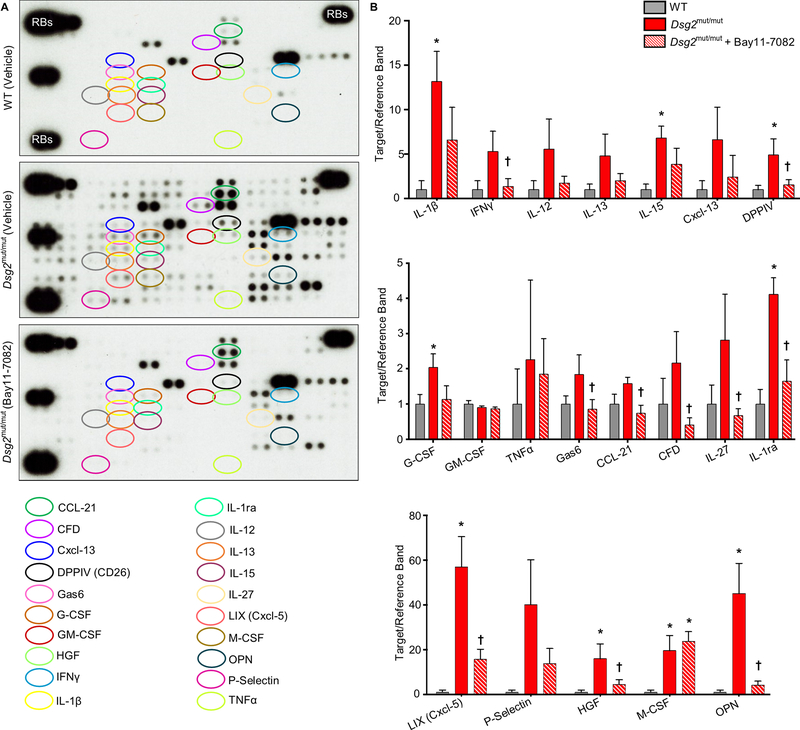
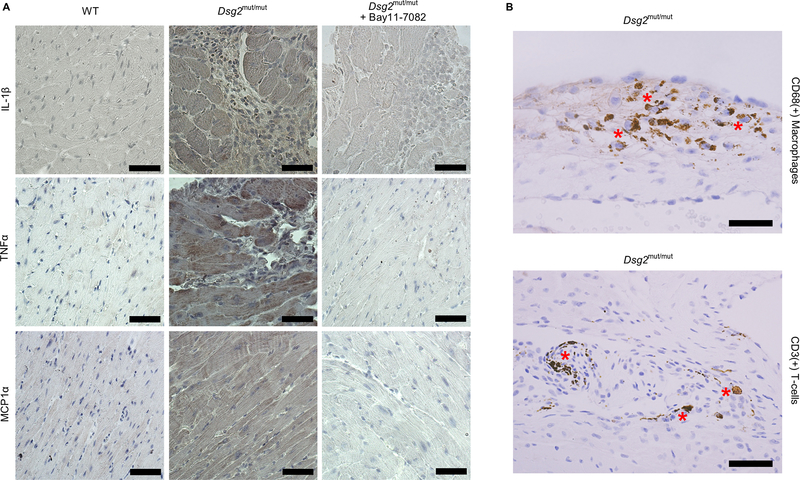
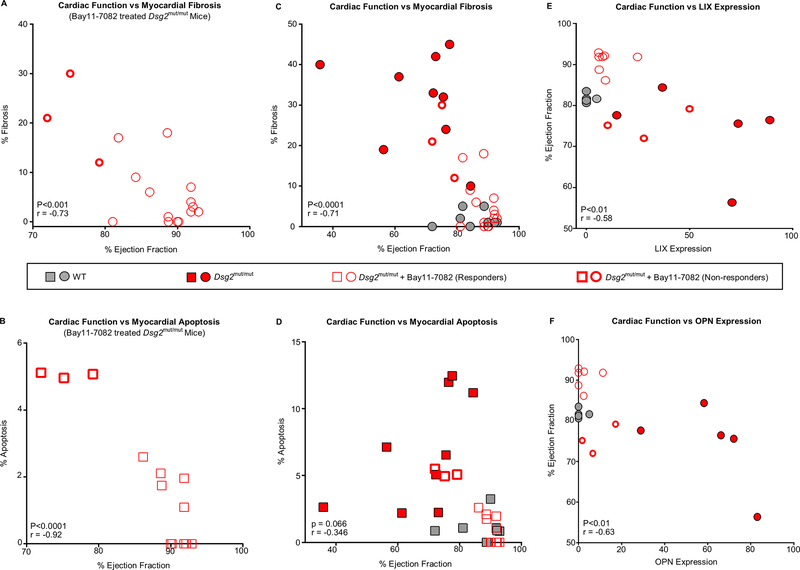
Results: Activation of nuclear factor-κB signaling, indicated by increased expression and nuclear accumulation of phospho-RelA/p65, occurred in both an in vitro model of ACM (expression of JUP2157del2 in neonatal rat ventricular myocytes) and a robust murine model of ACM (homozygous knock-in of mutant desmoglein-2 [Dsg2mut/mut]) that recapitulates the cardiac manifestations seen in patients with ACM. Bay 11-7082, a small-molecule inhibitor of nuclear factor-κB signaling, prevented the development of ACM disease features in vitro (abnormal redistribution of intercalated disk proteins, myocyte apoptosis, release of inflammatory cytokines) and in vivo (myocardial necrosis and fibrosis, left ventricular contractile dysfunction, electrocardiographic abnormalities). Hearts of Dsg2mut/mut mice expressed markedly increased levels of inflammatory cytokines and chemotactic molecules that were attenuated by Bay 11-7082. Salutary effects of Bay 11-7082 correlated with the extent to which production of selected cytokines had been blocked. Nuclear factor-κB signaling was also activated in cardiac myocytes derived from a patient with ACM. These cells produced and secreted abundant inflammatory cytokines under basal conditions, and this was also greatly reduced by Bay 11-7082.
Conclusions: Inflammatory signaling is activated in ACM and drives key features of the disease. Targeting inflammatory pathways may be an effective new mechanism-based therapy for ACM.
Keywords: B; NF-kappa; cardiomyopathies; cytokines; inflammation.
Conflict of interest statement
Conflict of Interest Disclosures
None
Figures

References
-
- Sen-Chowdhry S, Morgan RD, Chambers JC, McKenna WJ. Arrhythmogenic cardiomyopathy: etiology, diagnosis, and treatment. Annu Rev Med. 2010; 61:233–253. - PubMed
-
- Basso C, Bauce B, Corrado D, Thiene G. Pathophysiology of arrhythmogenic cardiomyopathy. Nat Rev Cardiol. 2012; 9:223–233. - PubMed
-
- Corrado D, Link M, Calkins H. Arrhythmogenic right ventricular cardiomyopathy. New Eng J Med. 2017; 376:61–72. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
