

Identification and characterization of NF1 and non-NF1 congenital pseudarthrosis of the tibia based on germline NF1 variants: genetic and clinical analysis of 75 patients
- PMID: 31533797
- PMCID: PMC6751843
- DOI: 10.1186/s13023-019-1196-0
Identification and characterization of NF1 and non-NF1 congenital pseudarthrosis of the tibia based on germline NF1 variants: genetic and clinical analysis of 75 patients
Abstract
Background: Congenital pseudarthrosis of the tibia (CPT) is a rare disease. Some patients present neurofibromatosis type 1 (NF1), while some others do not manifest NF1 (non-NF1). The etiology of CPT, particularly non-NF1 CPT, is not well understood. Here we screened germline variants of 75 CPT cases, including 55 NF1 and 20 non-NF1. Clinical data were classified and analyzed based on NF1 gene variations to investigate the genotype-phenotype relations of the two types of patients.
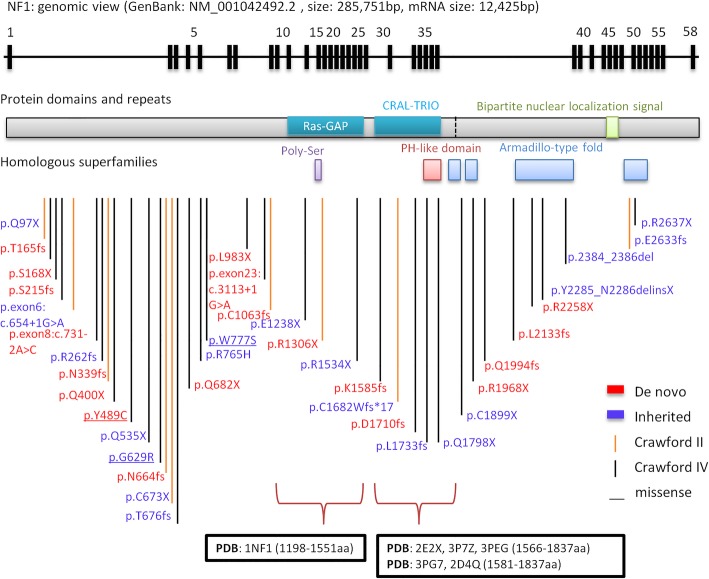
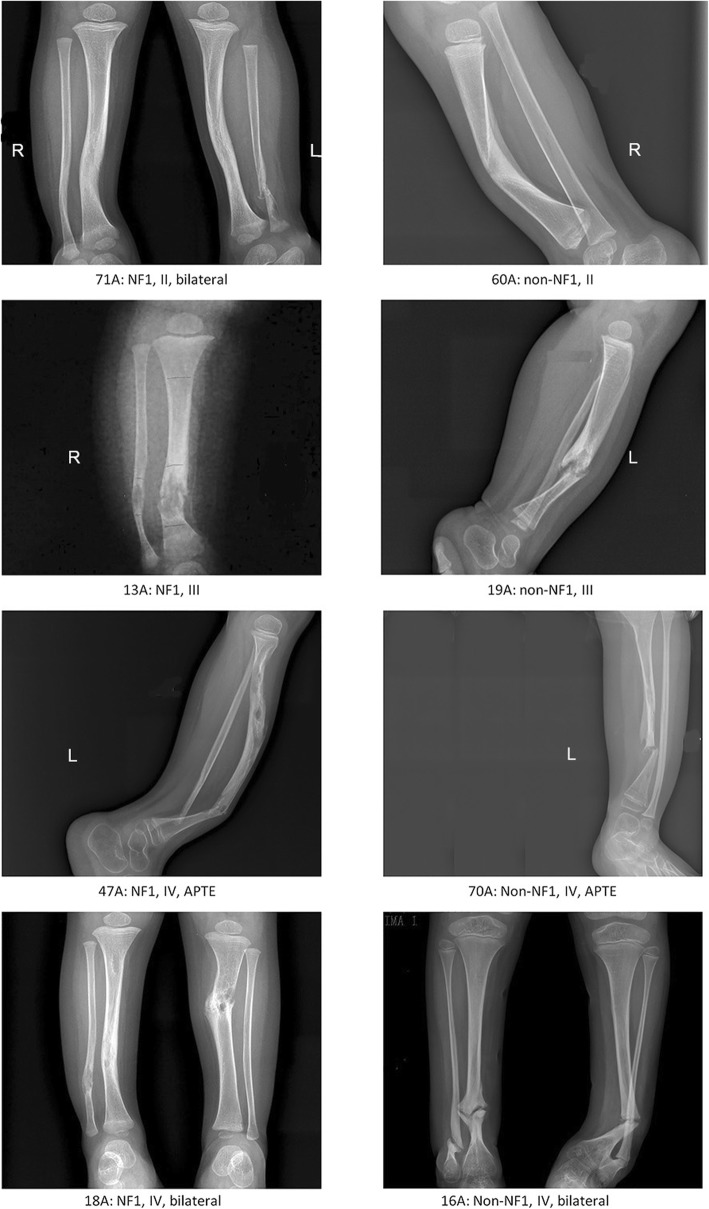
Results: Using whole-exome sequencing and Multiplex Ligation-Dependent Probe Amplification, 44 out of 55 NF1 CPT patients (80.0%) were identified as carrying pathogenic variants of the NF1 gene. Twenty-five variants were novel; 53.5% of variants were de novo, and a higher proportion of their carriers presented bone fractures compared to inherited variant carriers. No NF1 pathogenic variants were found in all 20 non-NF1 patients. Clinical features comparing NF1 CPT to non-NF1 CPT did not show significant differences in bowing or fracture onset, lateralization, tissue pathogenical results, abnormality of the proximal tibial epiphysis, and follow-up tibial union after surgery. A considerably higher proportion of non-NF1 patients have cystic lesion (Crawford type III) and used braces after surgery.
Conclusions: We analyzed a large cohort of non-NF1 and NF1 CPT patients and provided a new perspective for genotype-phenotype features related to germline NF1 variants. Non-NF1 CPT in general had similar clinical features of the tibia as NF1 CPT. Germline NF1 pathogenic variants could differentiate NF1 from non-NF1 CPT but could not explain the CPT heterogeneity of NF1 patients. Our results suggested that non-NF1 CPT was probably not caused by germline NF1 pathogenic variants. In addition to NF1, other genetic variants could also contribute to CPT pathogenesis. Our findings would facilitate the interpretation of NF1 pathogenic variants in CPT genetic counseling.
Keywords: Genomic variation; Genotype; Neurofibromatosis 1; Phenotype; Whole exome sequencing.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
-
- Crawford AH. Neurofibromatosis in children. Acta Orthop Scand Suppl. 1986;218:1–60. - PubMed
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
