

LTR retroelement expansion of the human cancer transcriptome and immunopeptidome revealed by de novo transcript assembly
- PMID: 31537638
- PMCID: PMC6771403
- DOI: 10.1101/gr.248922.119
LTR retroelement expansion of the human cancer transcriptome and immunopeptidome revealed by de novo transcript assembly
Abstract
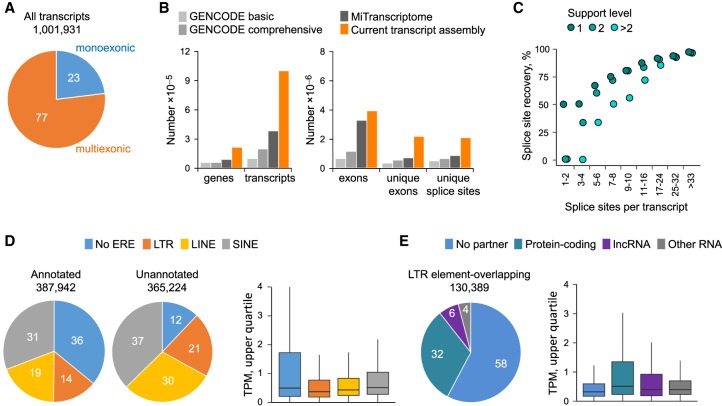
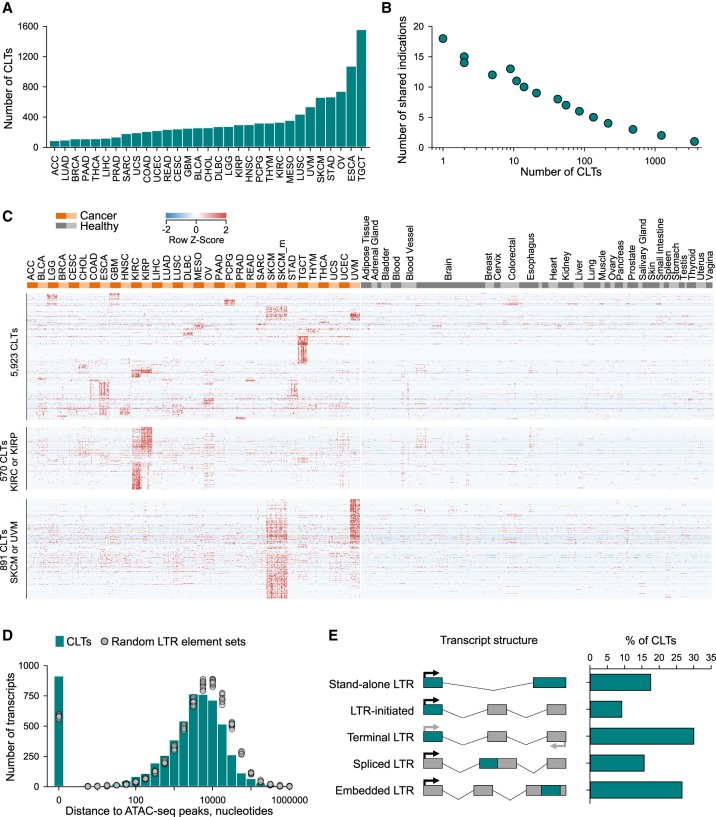
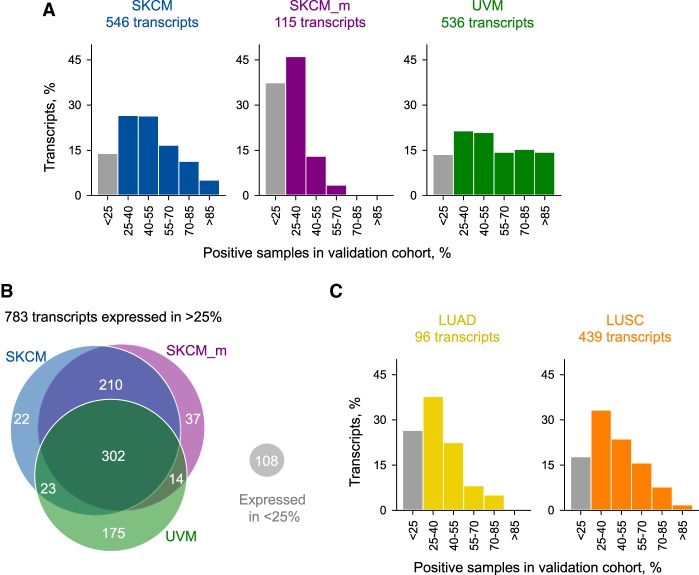
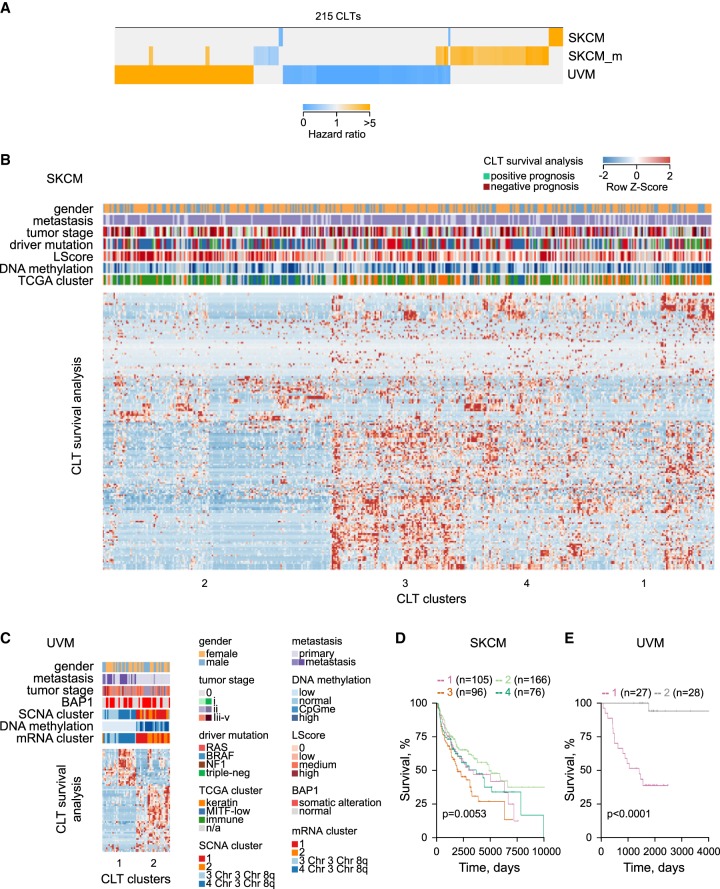
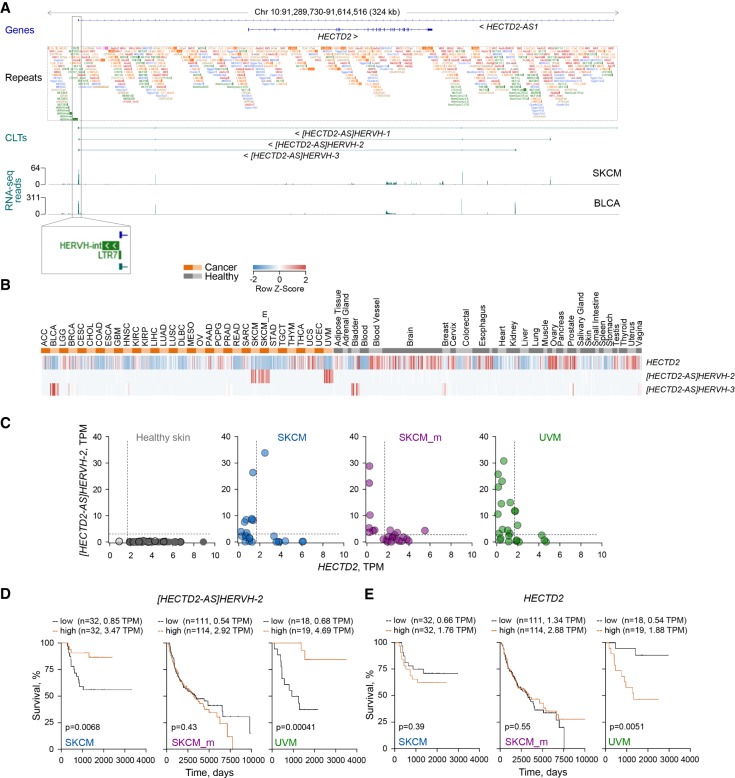
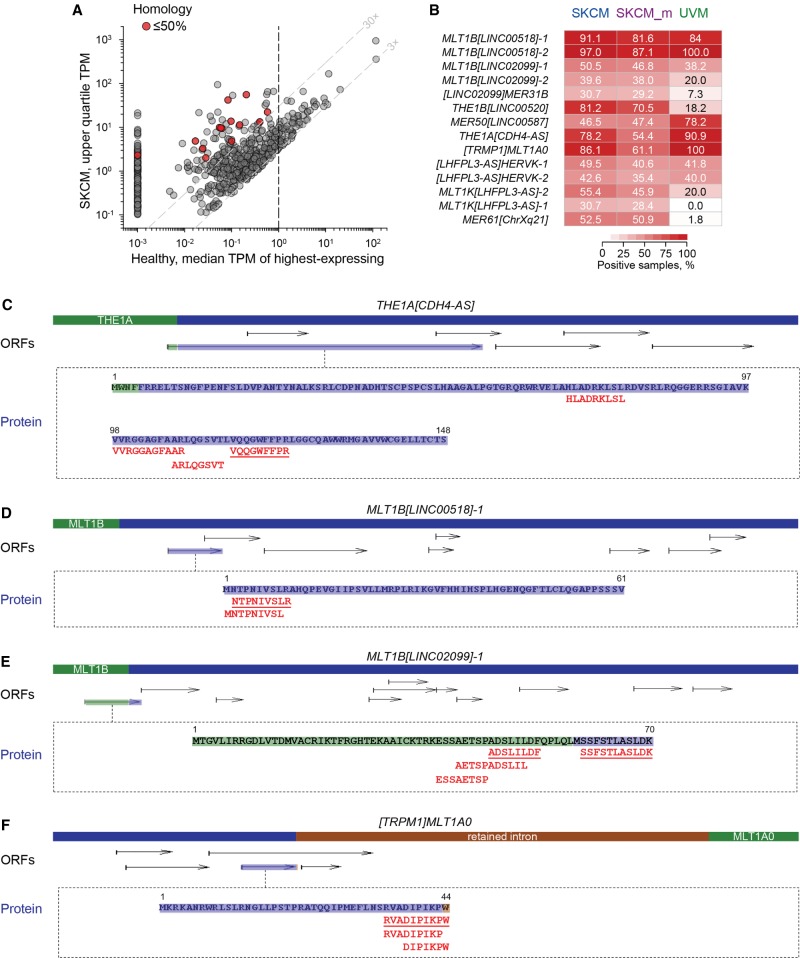
Dysregulated endogenous retroelements (EREs) are increasingly implicated in the initiation, progression, and immune surveillance of human cancer. However, incomplete knowledge of ERE activity limits mechanistic studies. By using pan-cancer de novo transcript assembly, we uncover the extent and complexity of ERE transcription. The current assembly doubled the number of previously annotated transcripts overlapping with long-terminal repeat (LTR) elements, several thousand of which were expressed specifically in one or a few related cancer types. Exemplified in melanoma, LTR-overlapping transcripts were highly predictable, disease prognostic, and closely linked with molecularly defined subtypes. They further showed the potential to affect disease-relevant genes, as well as produce novel cancer-specific antigenic peptides. This extended view of LTR elements provides the framework for functional validation of affected genes and targets for cancer immunotherapy.
© 2019 Attig et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Bassani-Sternberg M, Bräunlein E, Klar R, Engleitner T, Sinitcyn P, Audehm S, Straub M, Weber J, Slotta-Huspenina J, Specht K, et al. 2016. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat Commun 7: 13404 10.1038/ncomms13404 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources