

Concomitant PIK3CD and TNFRSF9 deficiencies cause chronic active Epstein-Barr virus infection of T cells
- PMID: 31537641
- PMCID: PMC6888974
- DOI: 10.1084/jem.20190678
Concomitant PIK3CD and TNFRSF9 deficiencies cause chronic active Epstein-Barr virus infection of T cells
Abstract
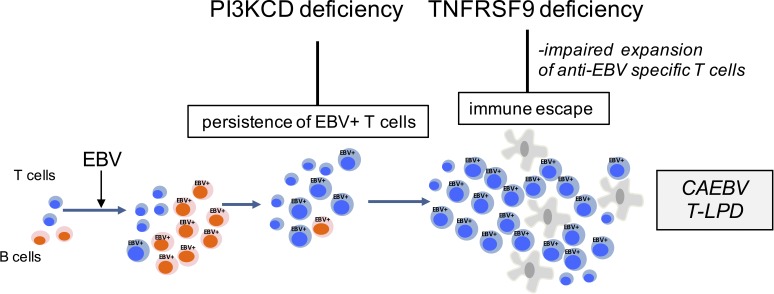
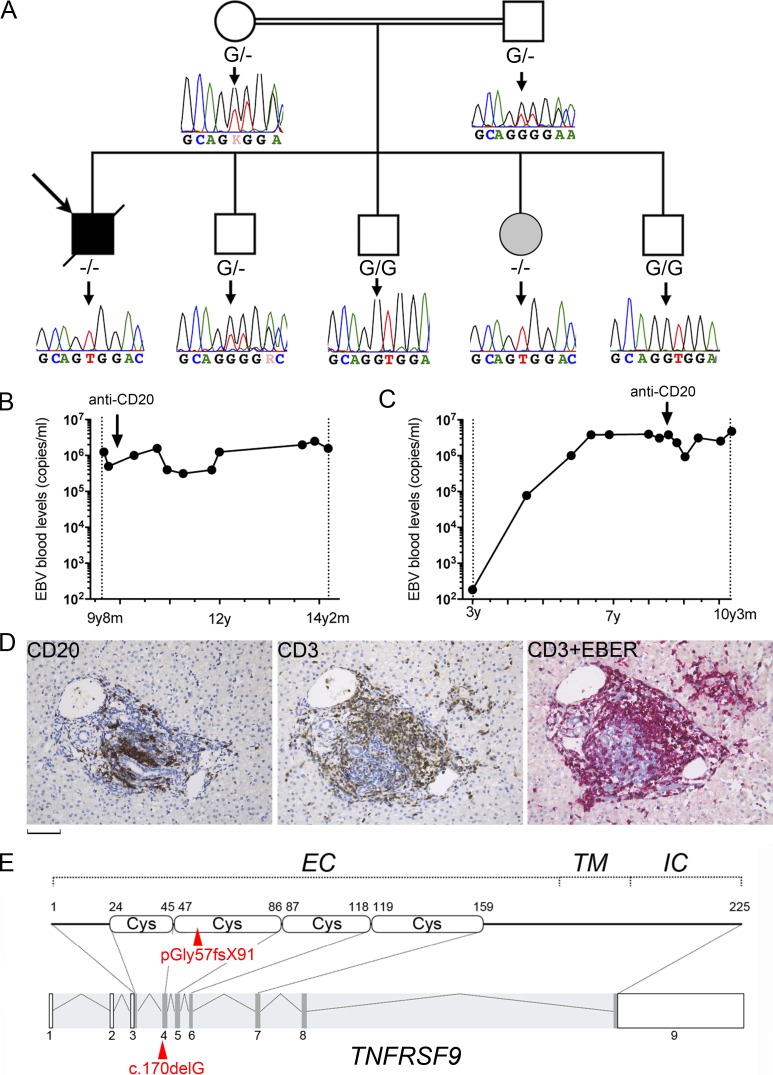
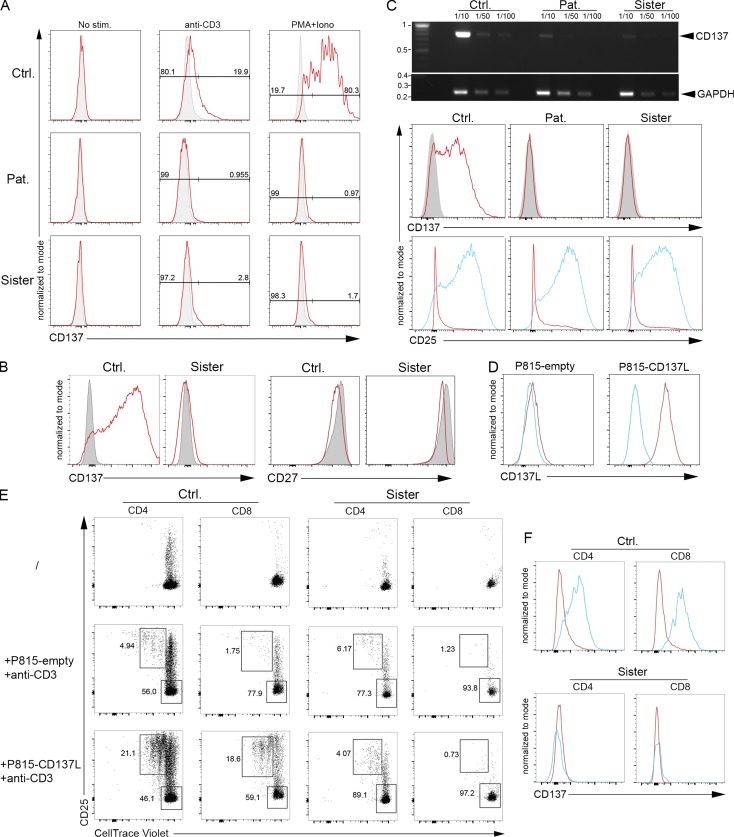
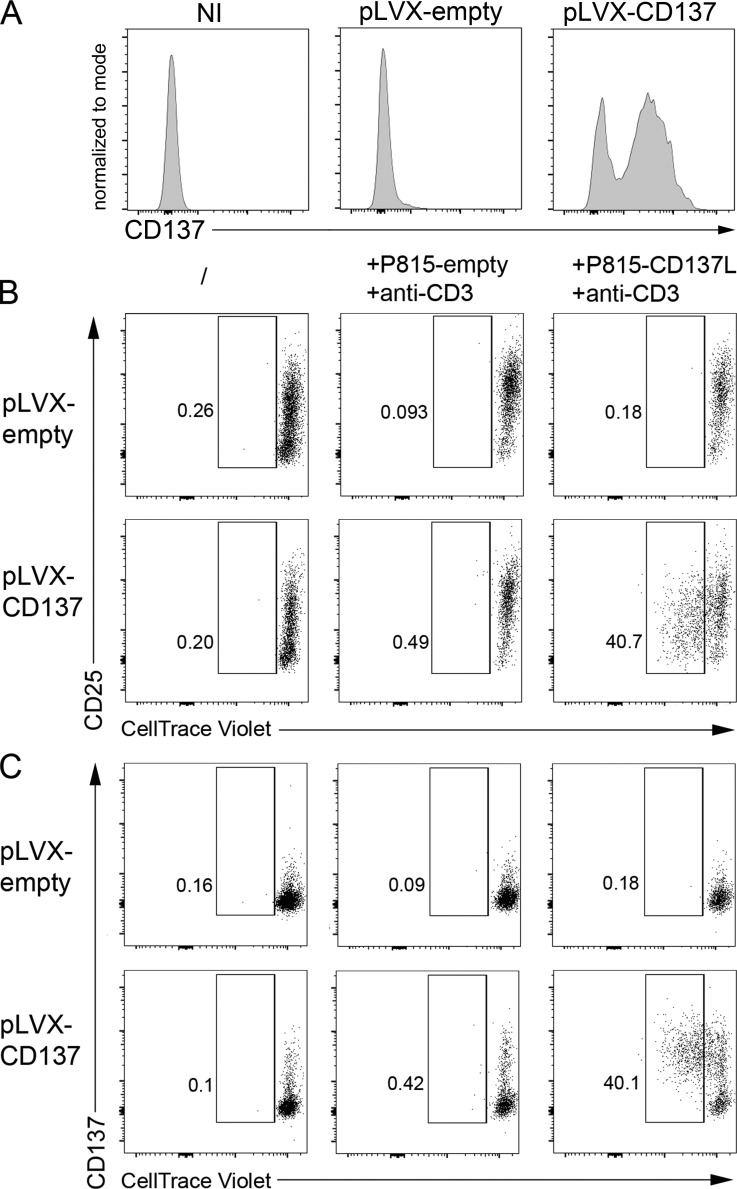
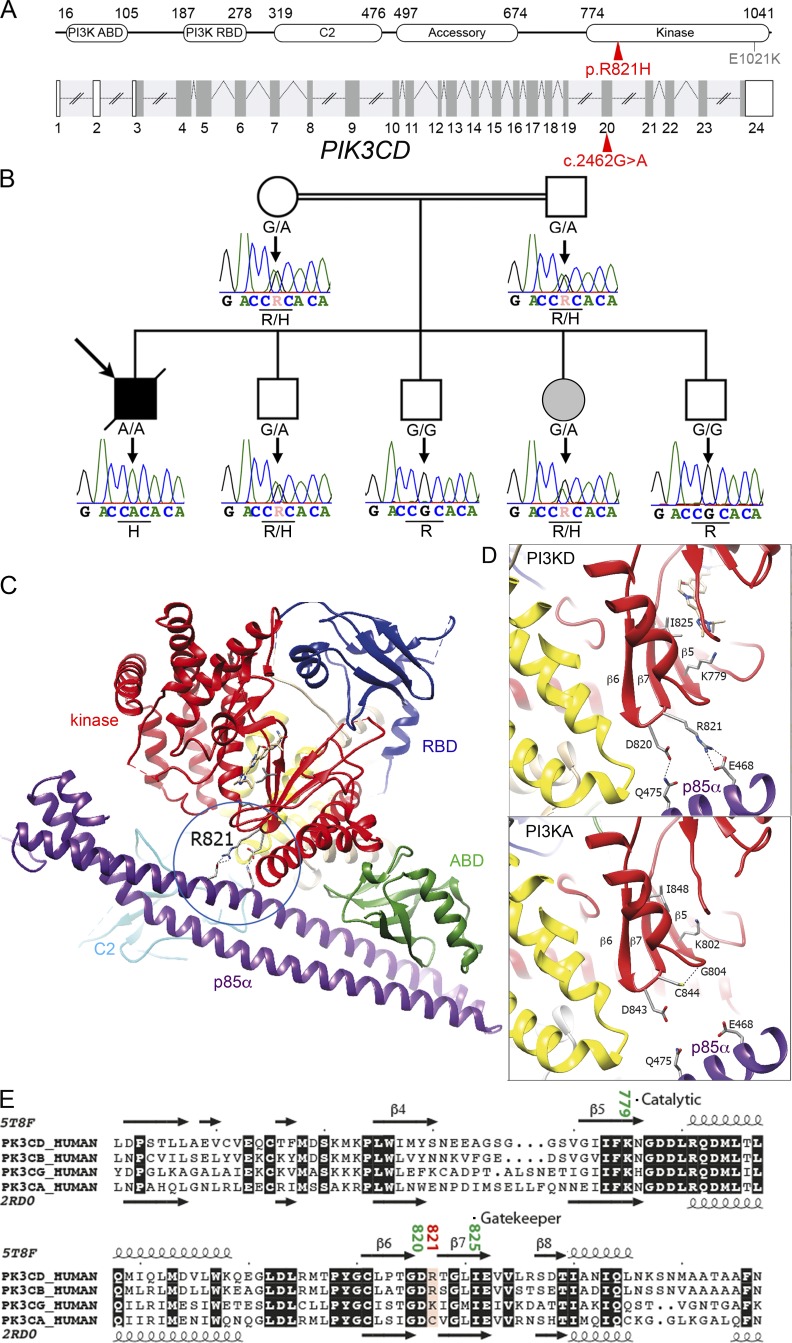
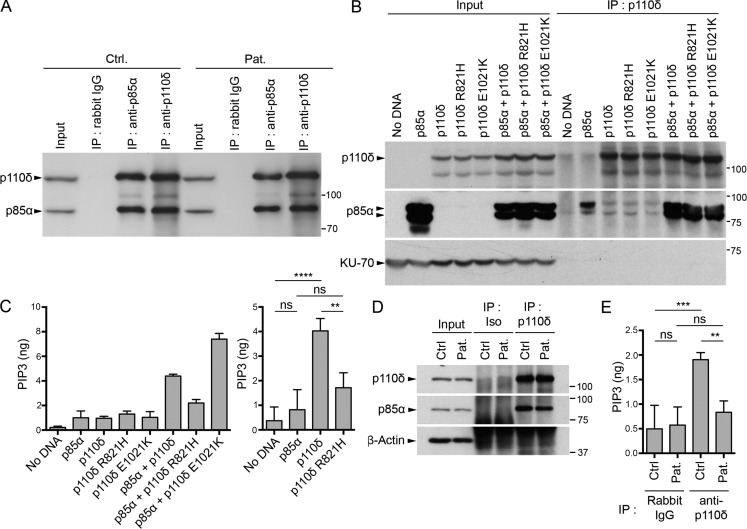
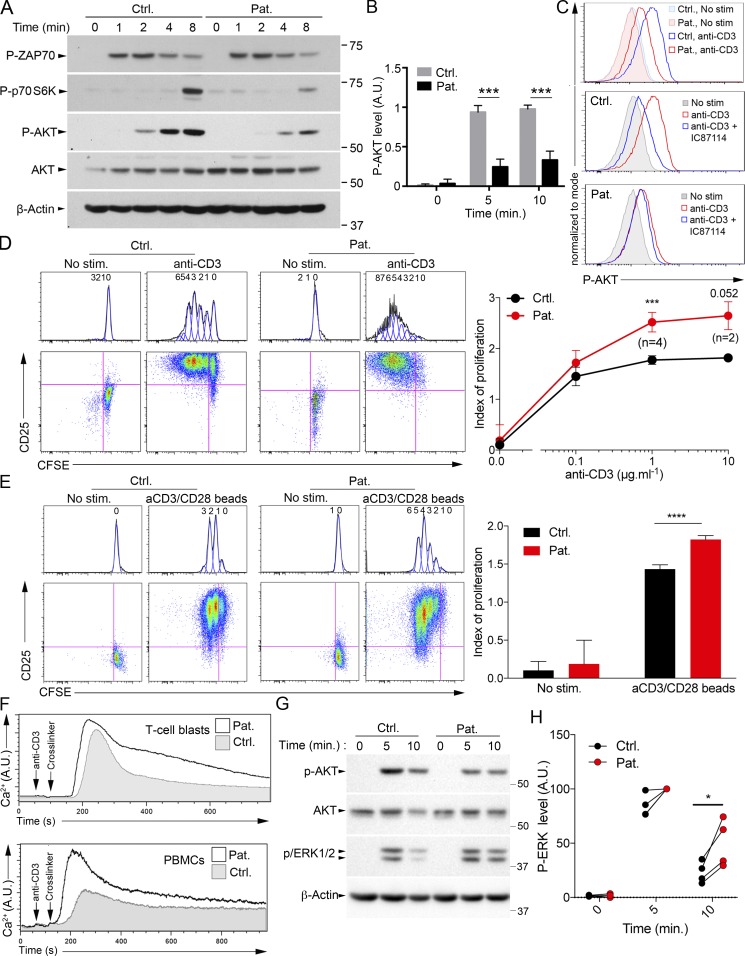
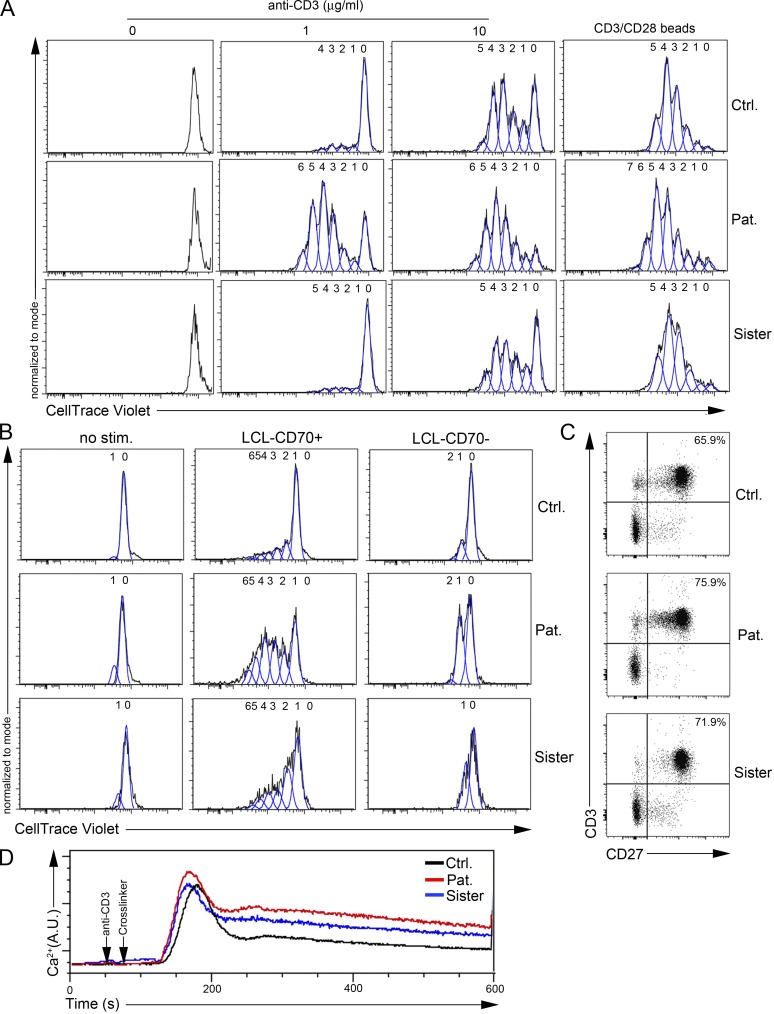
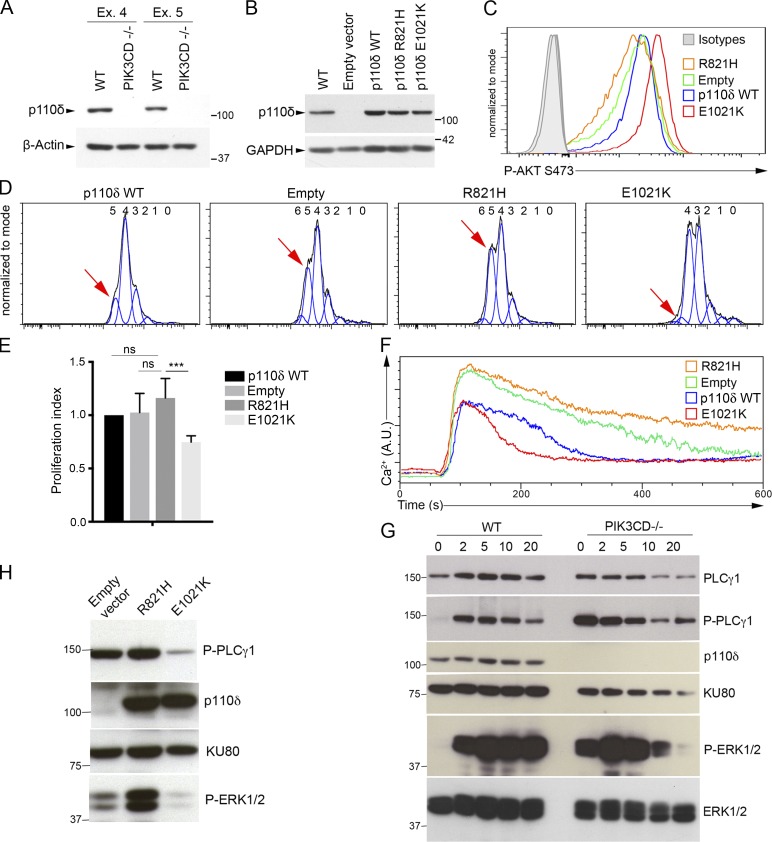
Infection of T cells by Epstein-Barr virus (EBV) causes chronic active EBV infection (CAEBV) characterized by T cell lymphoproliferative disorders (T-LPD) of unclear etiology. Here, we identified two homozygous biallelic loss-of-function mutations in PIK3CD and TNFRSF9 in a patient who developed a fatal CAEBV. The mutation in TNFRSF9 gene coding CD137/4-1BB, a costimulatory molecule expressed by antigen-specific activated T cells, resulted in a complete loss of CD137 expression and impaired T cell expansion toward CD137 ligand-expressing cells. Isolated as observed in one sibling, CD137 deficiency resulted in persistent EBV-infected T cells but without clinical manifestations. The mutation in PIK3CD gene that encodes the catalytic subunit p110δ of the PI3K significantly reduced its kinase activity. Deficient T cells for PIK3CD exhibited reduced AKT signaling, while calcium flux, RAS-MAPK activation, and proliferation were increased, suggestive of an imbalance between the PLCγ1 and PI3K pathways. These skewed signals in T cells may sustain accumulation of EBV-infected T cells, a process controlled by the CD137-CD137L pathway, highlighting its critical role in immunity to EBV.
© 2019 Rodriguez et al.
Figures
References
-
- Abolhassani, H., Edwards E.S., Ikinciogullari A., Jing H., Borte S., Buggert M., Du L., Matsuda-Lennikov M., Romano R., Caridha R., et al. . 2017. Combined immunodeficiency and Epstein-Barr virus-induced B cell malignancy in humans with inherited CD70 deficiency. J. Exp. Med. 214:91–106. 10.1084/jem.20160849 - DOI - PMC - PubMed
-
- Alkhairy, O.K., Perez-Becker R., Driessen G.J., Abolhassani H., van Montfrans J., Borte S., Choo S., Wang N., Tesselaar K., Fang M., et al. . 2015. Novel mutations in TNFRSF7/CD27: Clinical, immunologic, and genetic characterization of human CD27 deficiency. J. Allergy Clin. Immunol. 136:703–712.e10. 10.1016/j.jaci.2015.02.022 - DOI - PubMed
-
- Alosaimi, M.F., Hoenig M., Jaber F., Platt C.D., Jones J., Wallace J., Debatin K.M., Schulz A., Jacobsen E., Möller P., et al. . 2019. Immunodeficiency and EBV-induced lymphoproliferation caused by 4-1BB deficiency. J. Allergy Clin. Immunol. 144:574–583.e5. 10.1016/j.jaci.2019.03.002 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
