

Chromosome instability syndromes
- PMID: 31537806
- PMCID: PMC10617425
- DOI: 10.1038/s41572-019-0113-0
Chromosome instability syndromes
Abstract
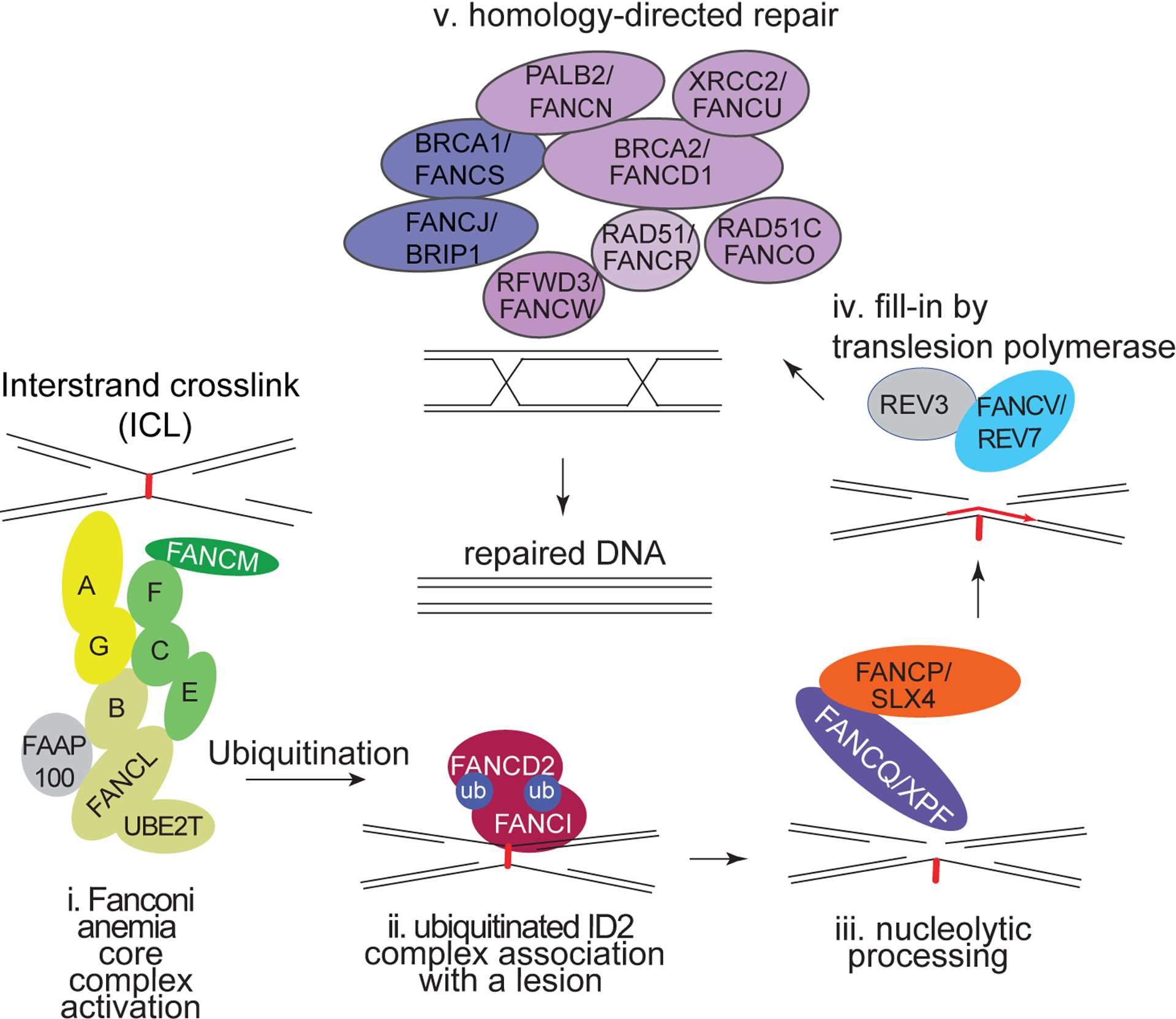
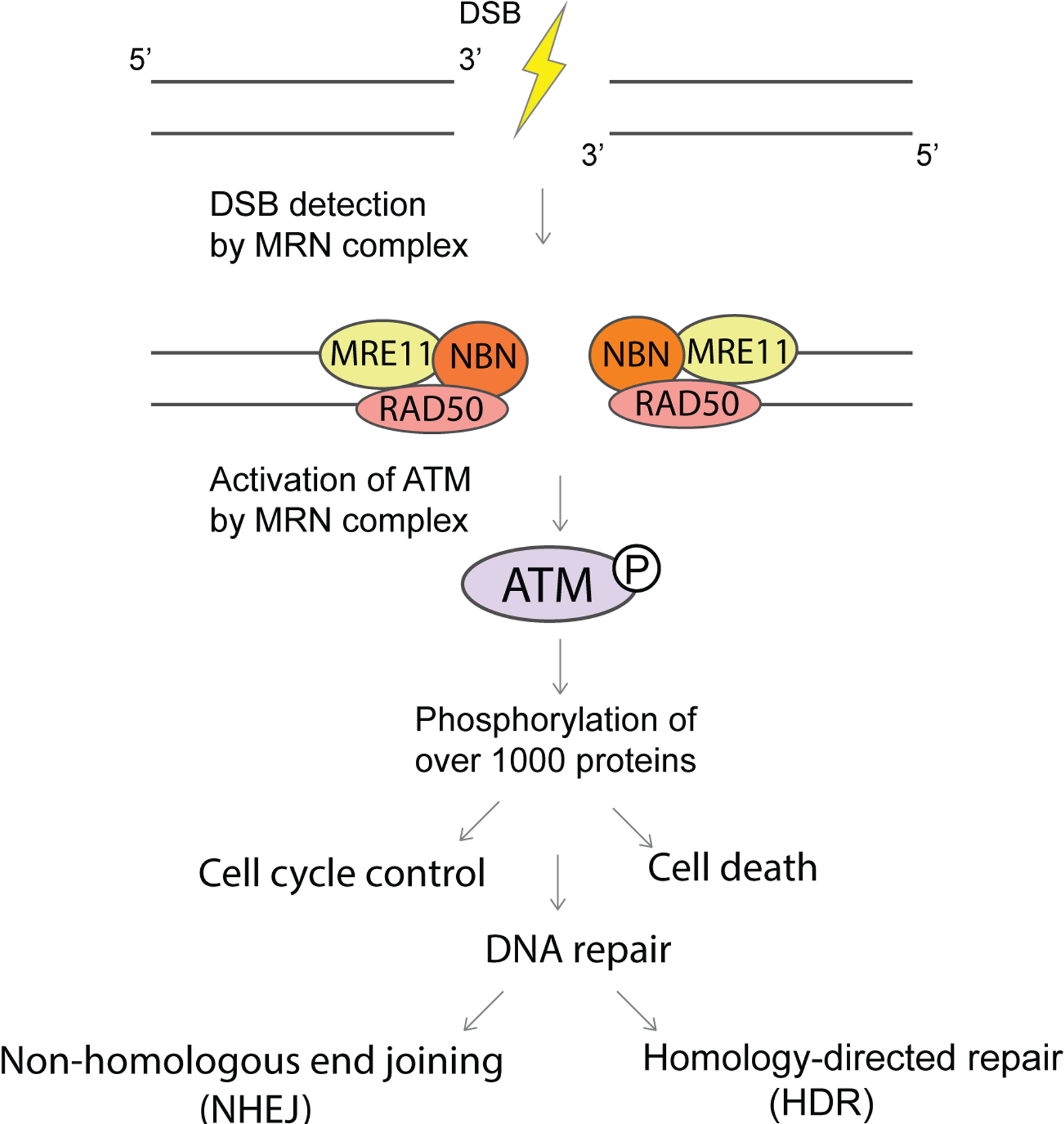
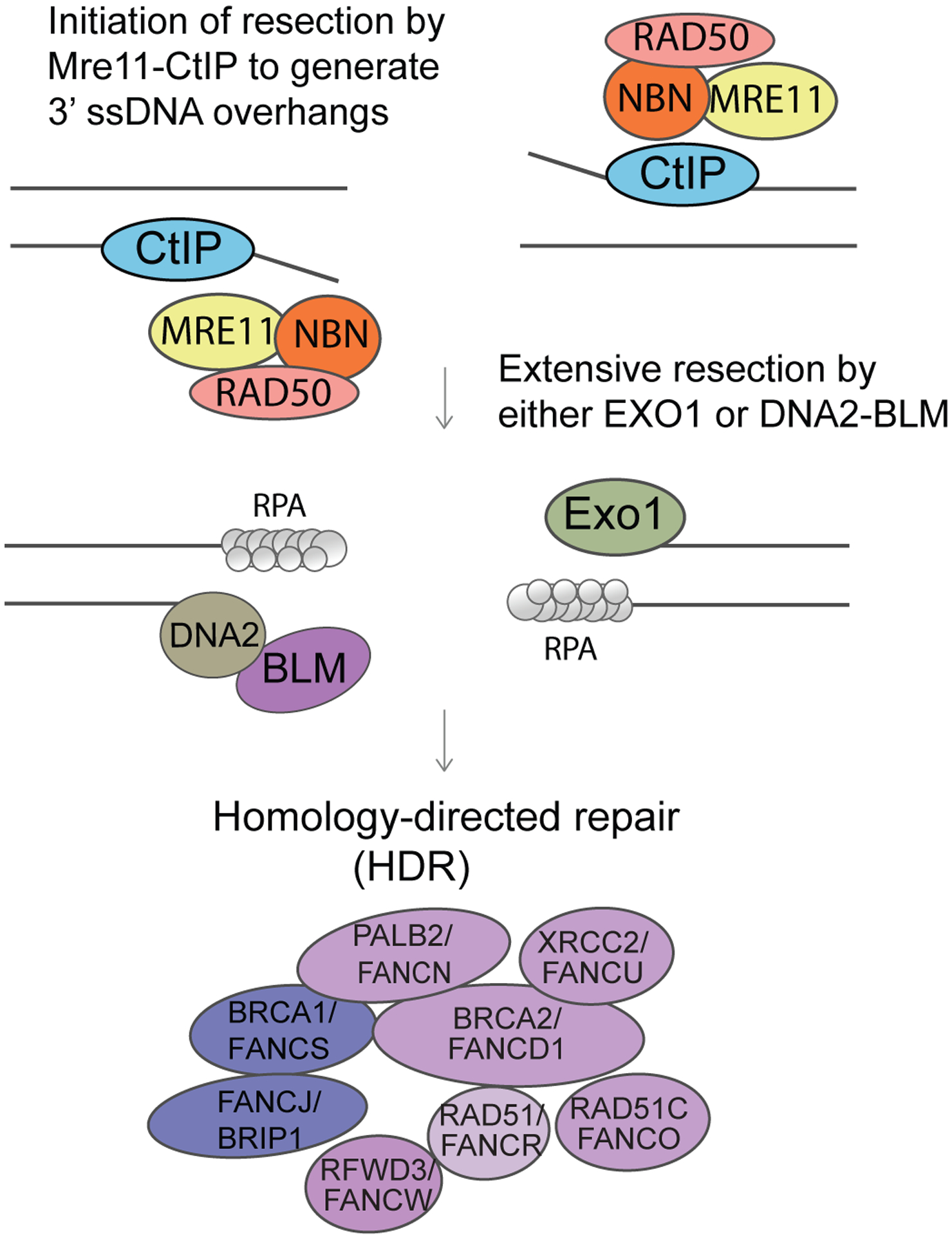
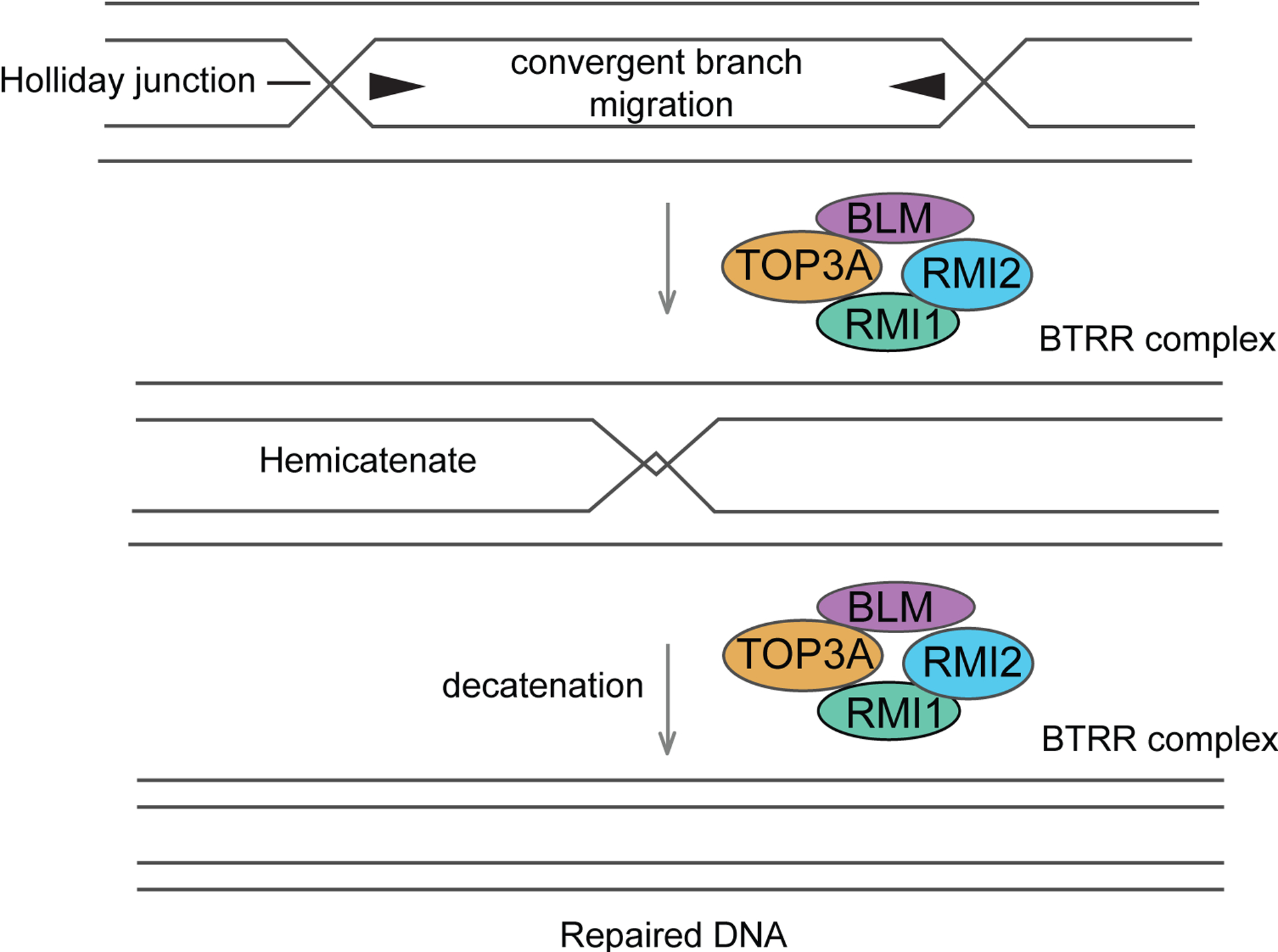
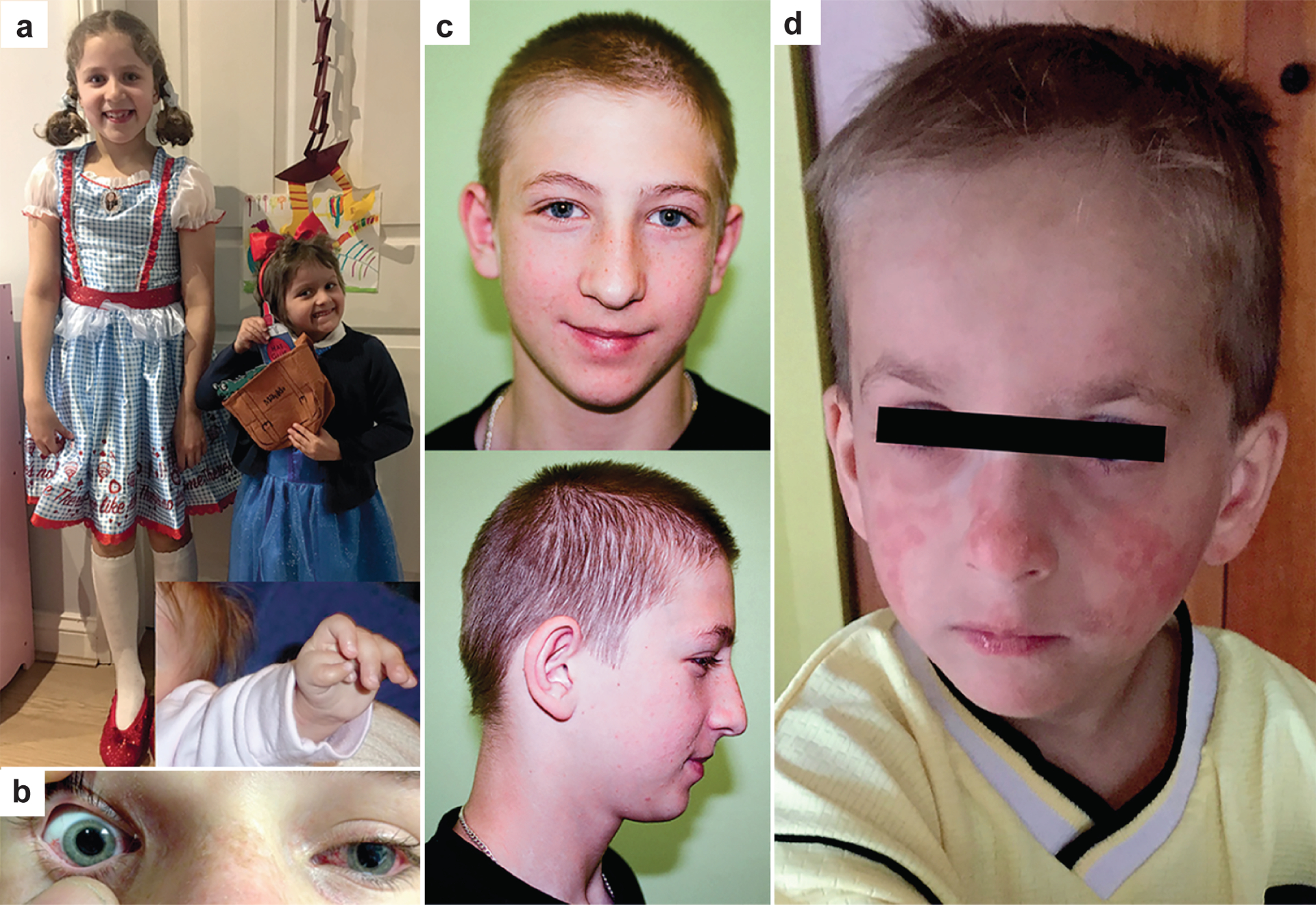
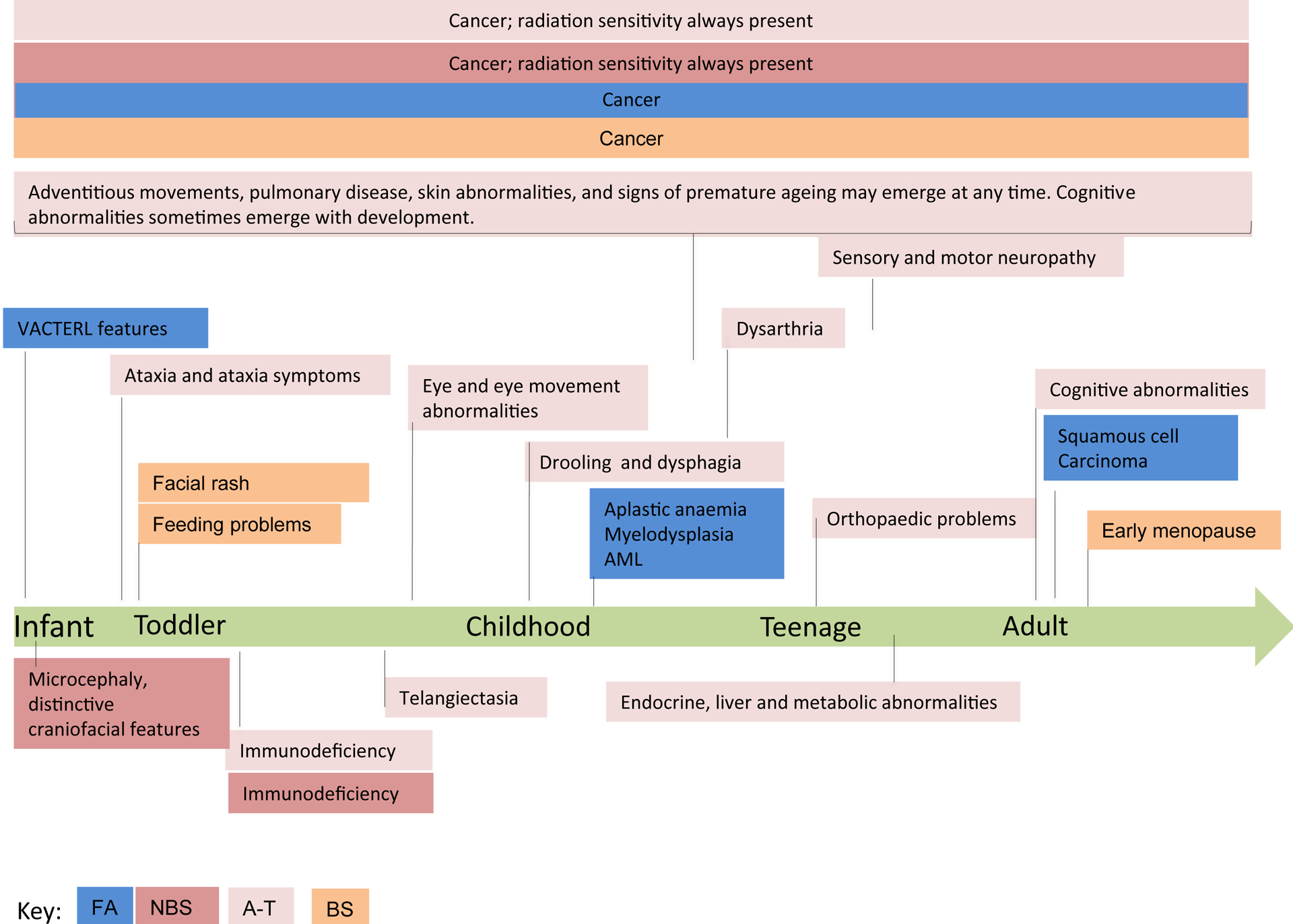
Fanconi anaemia (FA), ataxia telangiectasia (A-T), Nijmegen breakage syndrome (NBS) and Bloom syndrome (BS) are clinically distinct, chromosome instability (or breakage) disorders. Each disorder has its own pattern of chromosomal damage, with cells from these patients being hypersensitive to particular genotoxic drugs, indicating that the underlying defect in each case is likely to be different. In addition, each syndrome shows a predisposition to cancer. Study of the molecular and genetic basis of these disorders has revealed mechanisms of recognition and repair of DNA double-strand breaks, DNA interstrand crosslinks and DNA damage during DNA replication. Specialist clinics for each disorder have provided the concentration of expertise needed to tackle their characteristic clinical problems and improve outcomes. Although some treatments of the consequences of a disorder may be possible, for example, haematopoietic stem cell transplantation in FA and NBS, future early intervention to prevent complications of disease will depend on a greater understanding of the roles of the affected DNA repair pathways in development. An important realization has been the predisposition to cancer in carriers of some of these gene mutations.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
