

Yeast Models for Amyloids and Prions: Environmental Modulation and Drug Discovery
- PMID: 31540362
- PMCID: PMC6767215
- DOI: 10.3390/molecules24183388
Yeast Models for Amyloids and Prions: Environmental Modulation and Drug Discovery
Abstract
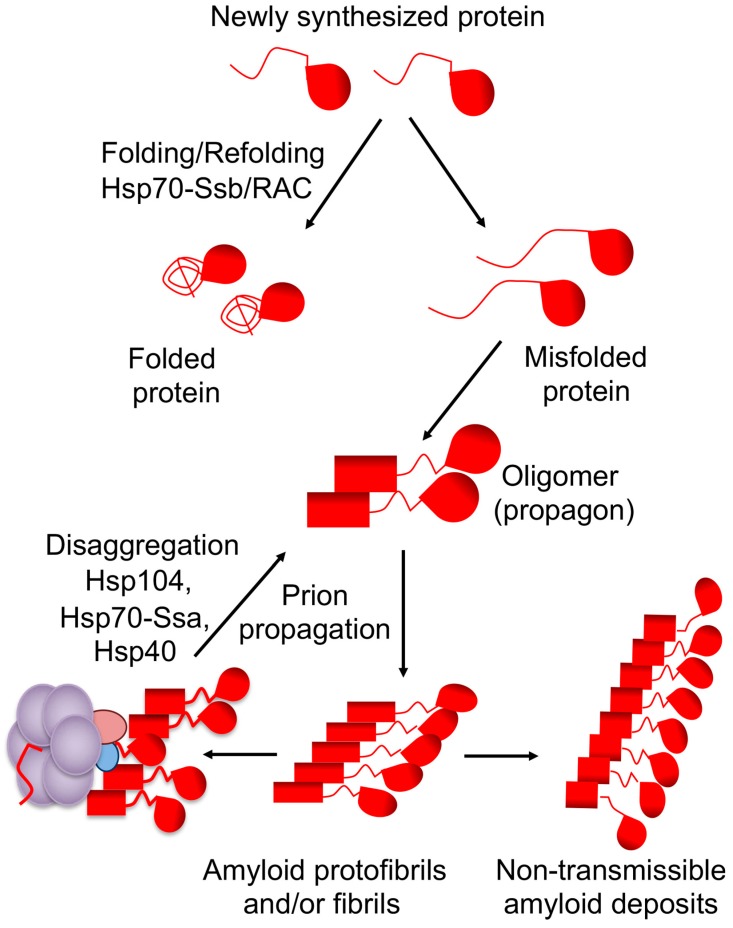
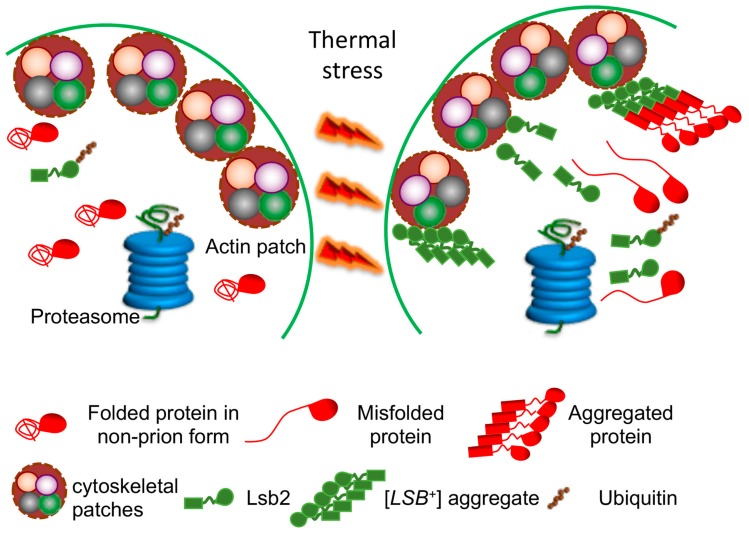
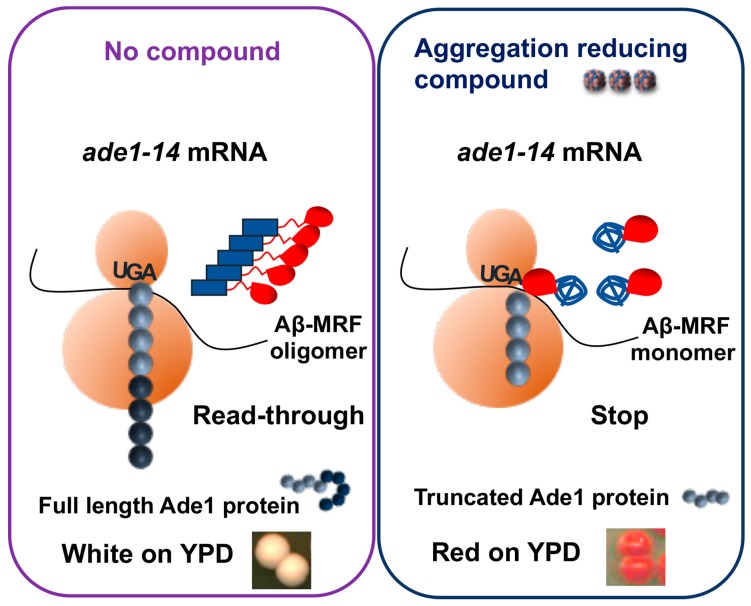
Amyloids are self-perpetuating protein aggregates causing neurodegenerative diseases in mammals. Prions are transmissible protein isoforms (usually of amyloid nature). Prion features were recently reported for various proteins involved in amyloid and neural inclusion disorders. Heritable yeast prions share molecular properties (and in the case of polyglutamines, amino acid composition) with human disease-related amyloids. Fundamental protein quality control pathways, including chaperones, the ubiquitin proteasome system and autophagy are highly conserved between yeast and human cells. Crucial cellular proteins and conditions influencing amyloids and prions were uncovered in the yeast model. The treatments available for neurodegenerative amyloid-associated diseases are few and their efficiency is limited. Yeast models of amyloid-related neurodegenerative diseases have become powerful tools for high-throughput screening for chemical compounds and FDA-approved drugs that reduce aggregation and toxicity of amyloids. Although some environmental agents have been linked to certain amyloid diseases, the molecular basis of their action remains unclear. Environmental stresses trigger amyloid formation and loss, acting either via influencing intracellular concentrations of the amyloidogenic proteins or via heterologous inducers of prions. Studies of environmental and physiological regulation of yeast prions open new possibilities for pharmacological intervention and/or prophylactic procedures aiming on common cellular systems rather than the properties of specific amyloids.
Keywords: amyloid; chaperone; drug discovery; environmental factors; heat shock; neurodegenerative disease; prion; ubiquitin.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
