

The bacterial endoribonuclease RNase E can cleave RNA in the absence of the RNA chaperone Hfq
- PMID: 31540970
- PMCID: PMC6827277
- DOI: 10.1074/jbc.RA119.010105
The bacterial endoribonuclease RNase E can cleave RNA in the absence of the RNA chaperone Hfq
Abstract
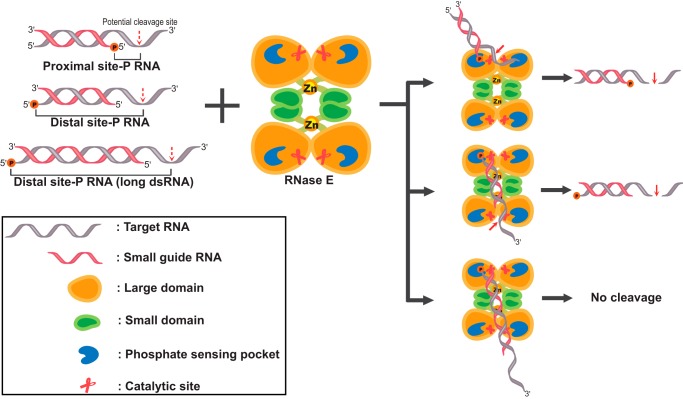
RNase E is a component of the RNA degradosome complex and plays a key role in RNA degradation and maturation in Escherichia coli RNase E-mediated target RNA degradation typically involves the RNA chaperone Hfq and requires small guide RNAs (sRNAs) acting as a seed by binding to short (7-12-bp) complementary regions in target RNA sequences. Here, using recombinantly expressed and purified proteins, site-directed mutagenesis, and RNA cleavage and protein cross-linking assays, we investigated Hfq-independent RNA decay by RNase E. Exploring its RNA substrate preferences in the absence of Hfq, we observed that RNase E preferentially cleaves AU-rich sites of single-stranded regions of RNA substrates that are annealed to an sRNA that contains a monophosphate at its 5'-end. We further found that the quaternary structure of RNase E is also important for complete, Hfq-independent cleavage at sites both proximal and distal to the sRNA-binding site within target RNAs containing monophosphorylated 5'-ends. Of note, genetic RNase E variants with unstable quaternary structure exhibited decreased catalytic activity. In summary, our results show that RNase E can degrade its target RNAs in the absence of the RNA chaperone Hfq. We conclude that RNase E-mediated, Hfq-independent RNA decay in E. coli requires a cognate sRNA sequence for annealing to the target RNA, a 5'-monophosphate at the RNA 5'-end, and a stable RNase E quaternary structure.
Keywords: RNA chaperone Hfq; RNA degradation; RNA processing; RNA turnover; RNase E; endoribonuclease; mRNA decay; protein expression; ribonuclease; small guide RNA (sRNA).
© 2019 Baek et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures






Similar articles
-
Dynamic interactions between the RNA chaperone Hfq, small regulatory RNAs, and mRNAs in live bacterial cells.Elife. 2021 Feb 22;10:e64207. doi: 10.7554/eLife.64207. Elife. 2021. PMID: 33616037 Free PMC article.
-
The seed region of a small RNA drives the controlled destruction of the target mRNA by the endoribonuclease RNase E.Mol Cell. 2012 Sep 28;47(6):943-53. doi: 10.1016/j.molcel.2012.07.015. Epub 2012 Aug 16. Mol Cell. 2012. PMID: 22902561 Free PMC article.
-
Acidic Residues in the Hfq Chaperone Increase the Selectivity of sRNA Binding and Annealing.J Mol Biol. 2015 Nov 6;427(22):3491-3500. doi: 10.1016/j.jmb.2015.07.010. Epub 2015 Jul 18. J Mol Biol. 2015. PMID: 26196441 Free PMC article.
-
Bacterial small RNA-based negative regulation: Hfq and its accomplices.J Biol Chem. 2013 Mar 22;288(12):7996-8003. doi: 10.1074/jbc.R112.441386. Epub 2013 Jan 29. J Biol Chem. 2013. PMID: 23362267 Free PMC article. Review.
-
Structure and RNA-binding properties of the bacterial LSm protein Hfq.RNA Biol. 2013 Apr;10(4):610-8. doi: 10.4161/rna.24201. Epub 2013 Mar 27. RNA Biol. 2013. PMID: 23535768 Free PMC article. Review.
Cited by
-
Post-transcriptional regulation of redox homeostasis by the small RNA SHOxi in haloarchaea.RNA Biol. 2021 Nov;18(11):1867-1881. doi: 10.1080/15476286.2021.1874717. Epub 2021 Jan 31. RNA Biol. 2021. PMID: 33522404 Free PMC article.
-
Small RNAs Asserting Big Roles in Mycobacteria.Noncoding RNA. 2021 Oct 29;7(4):69. doi: 10.3390/ncrna7040069. Noncoding RNA. 2021. PMID: 34842799 Free PMC article. Review.
-
Substrate-dependent effects of quaternary structure on RNase E activity.Genes Dev. 2021 Feb 1;35(3-4):286-299. doi: 10.1101/gad.335828.119. Epub 2021 Jan 14. Genes Dev. 2021. PMID: 33446571 Free PMC article.
-
Nucleic acid-binding KH domain proteins influence a spectrum of biological pathways including as part of membrane-localized complexes.J Struct Biol X. 2024 Jun 27;10:100106. doi: 10.1016/j.yjsbx.2024.100106. eCollection 2024 Dec. J Struct Biol X. 2024. PMID: 39040530 Free PMC article.
-
Kinetic modeling reveals additional regulation at co-transcriptional level by post-transcriptional sRNA regulators.Cell Rep. 2021 Sep 28;36(13):109764. doi: 10.1016/j.celrep.2021.109764. Cell Rep. 2021. PMID: 34592145 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
