

SeqSero2: Rapid and Improved Salmonella Serotype Determination Using Whole-Genome Sequencing Data
- PMID: 31540993
- PMCID: PMC6856333
- DOI: 10.1128/AEM.01746-19
SeqSero2: Rapid and Improved Salmonella Serotype Determination Using Whole-Genome Sequencing Data
Abstract
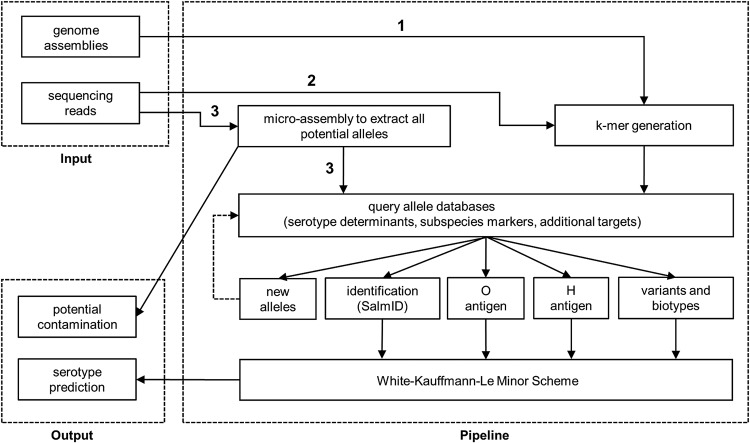
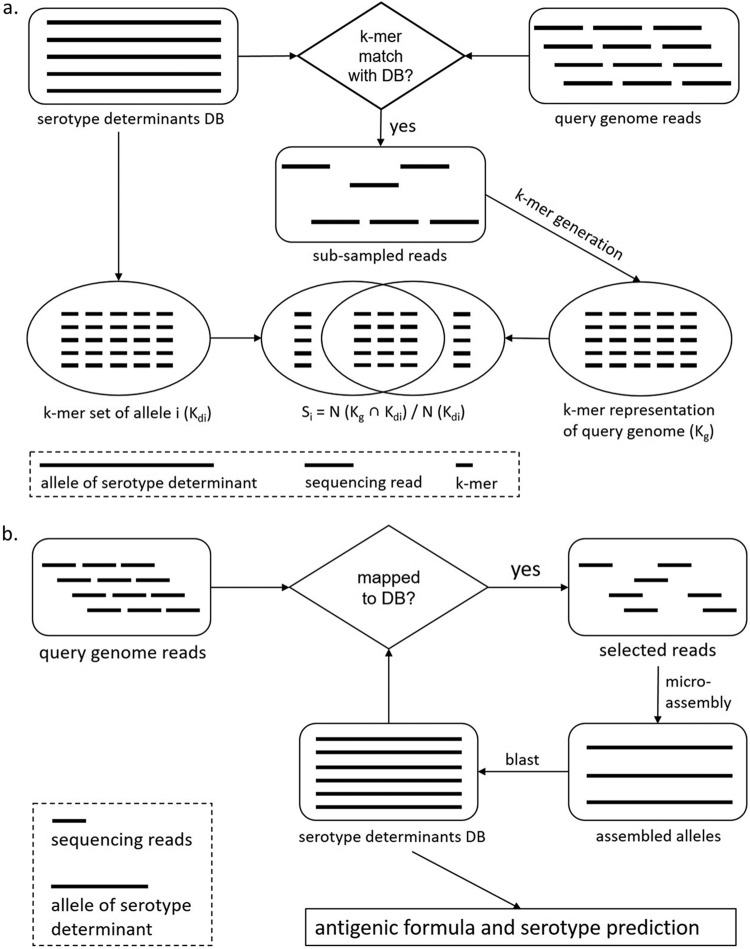
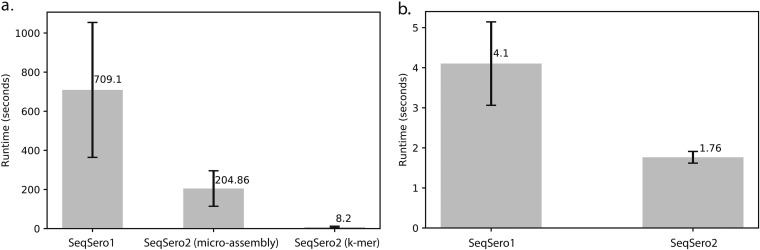
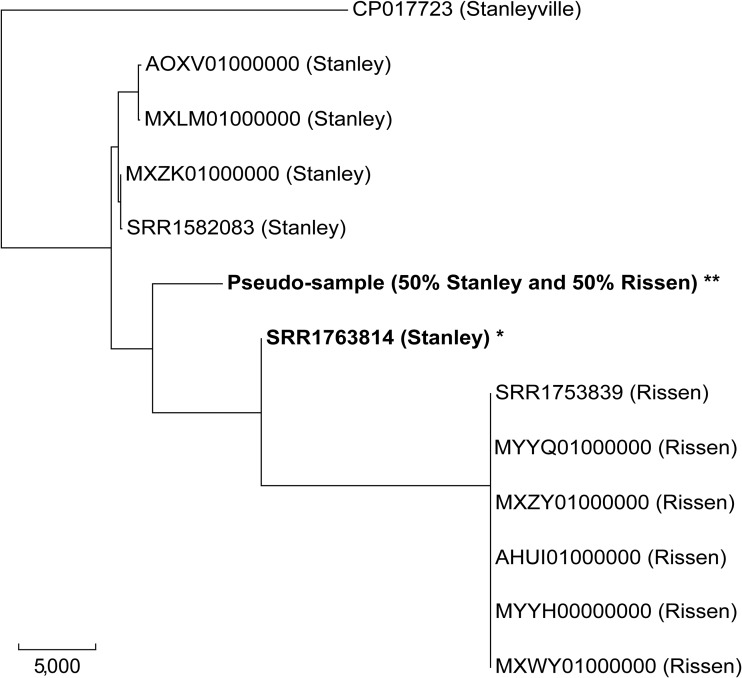
SeqSero, launched in 2015, is a software tool for Salmonella serotype determination from whole-genome sequencing (WGS) data. Despite its routine use in public health and food safety laboratories in the United States and other countries, the original SeqSero pipeline is relatively slow (minutes per genome using sequencing reads), is not optimized for draft genome assemblies, and may assign multiple serotypes for a strain. Here, we present SeqSero2 (github.com/denglab/SeqSero2; denglab.info/SeqSero2), an algorithmic transformation and functional update of the original SeqSero. Major improvements include (i) additional sequence markers for identification of Salmonella species and subspecies and certain serotypes, (ii) a k-mer based algorithm for rapid serotype prediction from raw reads (seconds per genome) and improved serotype prediction from assemblies, and (iii) a targeted assembly approach for specific retrieval of serotype determinants from WGS for serotype prediction, new allele discovery, and prediction troubleshooting. Evaluated using 5,794 genomes representing 364 common U.S. serotypes, including 2,280 human isolates of 117 serotypes from the National Antimicrobial Resistance Monitoring System, SeqSero2 is up to 50 times faster than the original SeqSero while maintaining equivalent accuracy for raw reads and substantially improving accuracy for assemblies. SeqSero2 further suggested that 3% of the tested genomes contained reads from multiple serotypes, indicating a use for contamination detection. In addition to short reads, SeqSero2 demonstrated potential for accurate and rapid serotype prediction directly from long nanopore reads despite base call errors. Testing of 40 nanopore-sequenced genomes of 17 serotypes yielded a single H antigen misidentification.IMPORTANCE Serotyping is the basis of public health surveillance of Salmonella It remains a first-line subtyping method even as surveillance continues to be transformed by whole-genome sequencing. SeqSero allows the integration of Salmonella serotyping into a whole-genome-sequencing-based laboratory workflow while maintaining continuity with the classic serotyping scheme. SeqSero2, informed by extensive testing and application of SeqSero in the United States and other countries, incorporates important improvements and updates that further strengthen its application in routine and large-scale surveillance of Salmonella by whole-genome sequencing.
Keywords: Salmonella; WGS; serotype; whole-genome sequencing.
Copyright © 2019 American Society for Microbiology.
Figures

References
-
- Ashton PM, Nair S, Peters TM, Bale JA, Powell DG, Painset A, Tewolde R, Schaefer U, Jenkins C, Dallman TJ, de Pinna EM, Grant KA, Salmonella Whole Genome Sequencing Implementation Group . 2016. Identification of Salmonella for public health surveillance using whole genome sequencing. PeerJ 4:e1752. doi:10.7717/peerj.1752. - DOI - PMC - PubMed
-
- Inns T, Ashton PM, Herrera-Leon S, Lighthill J, Foulkes S, Jombart T, Rehman Y, Fox A, Dallman T, DE Pinna E, Browning L, Coia JE, Edeghere O, Vivancos R. 2017. Prospective use of whole genome sequencing (WGS) detected a multi-country outbreak of Salmonella Enteritidis. Epidemiol Infect 145:289–298. doi:10.1017/S0950268816001941. - DOI - PMC - PubMed
-
- Grimont PAD, Weill F-X. 2007. Antigenic formulae of the Salmonella serovars, 9th ed WHO Collaborating Centre for Reference and Research on Salmonella, Paris, France.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
