

Removal of the Potassium Chloride Co-Transporter from the Somatodendritic Membrane of Axotomized Motoneurons Is Independent of BDNF/TrkB Signaling But Is Controlled by Neuromuscular Innervation
- PMID: 31541001
- PMCID: PMC6795555
- DOI: 10.1523/ENEURO.0172-19.2019
Removal of the Potassium Chloride Co-Transporter from the Somatodendritic Membrane of Axotomized Motoneurons Is Independent of BDNF/TrkB Signaling But Is Controlled by Neuromuscular Innervation
Abstract
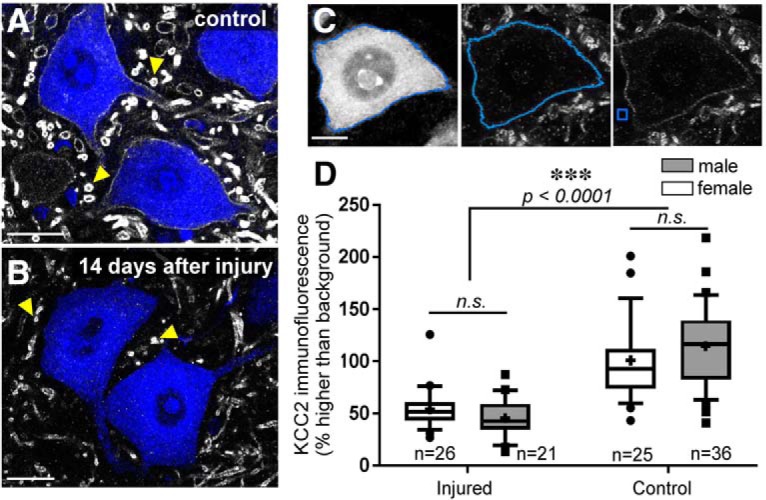
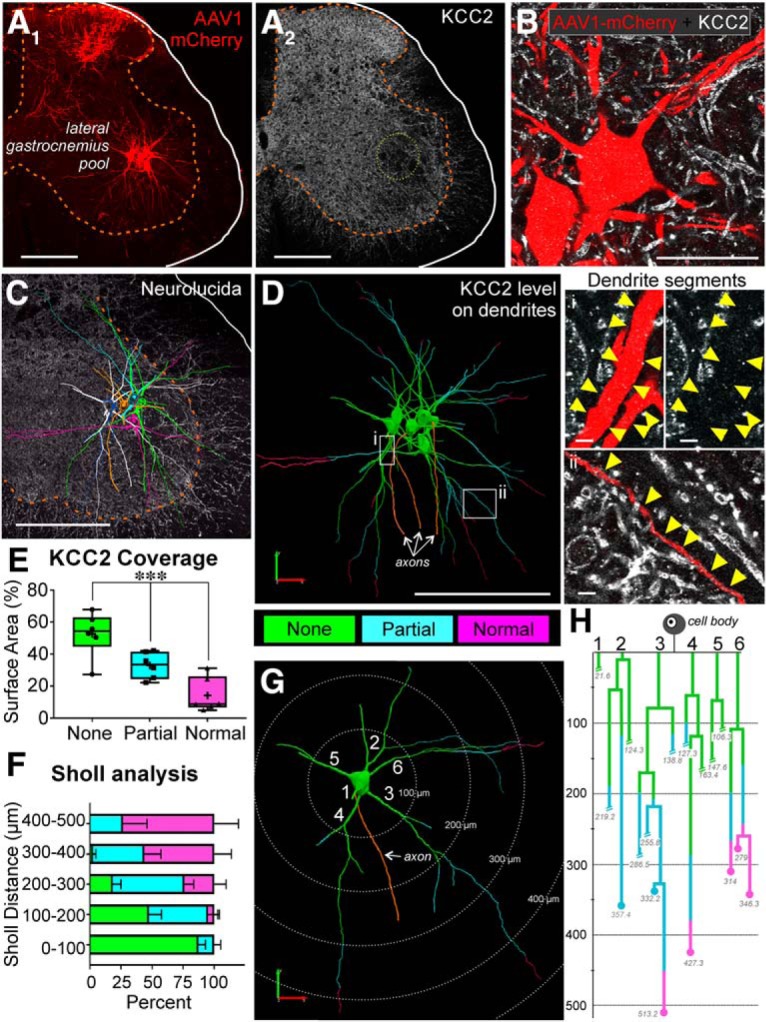
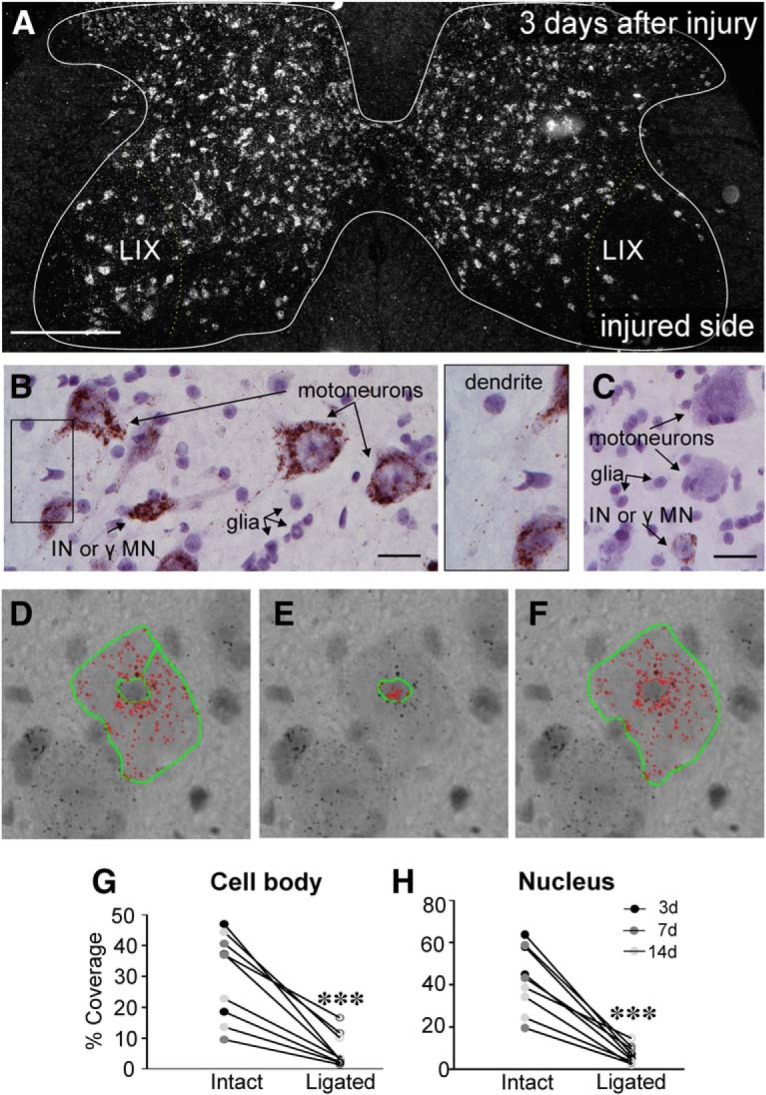
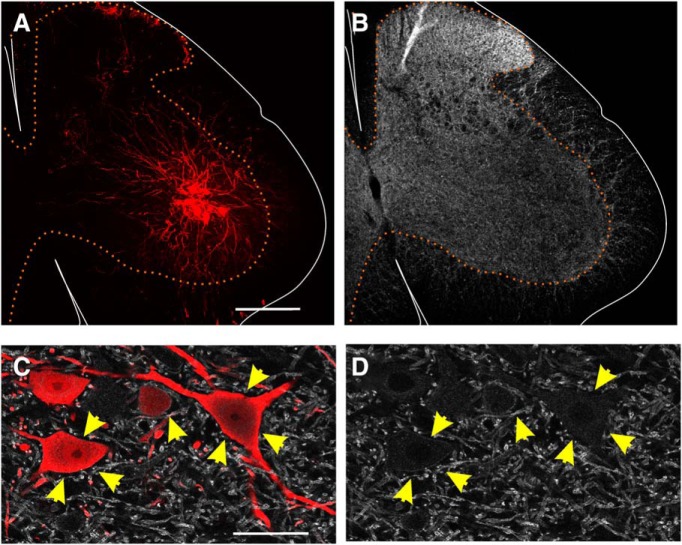
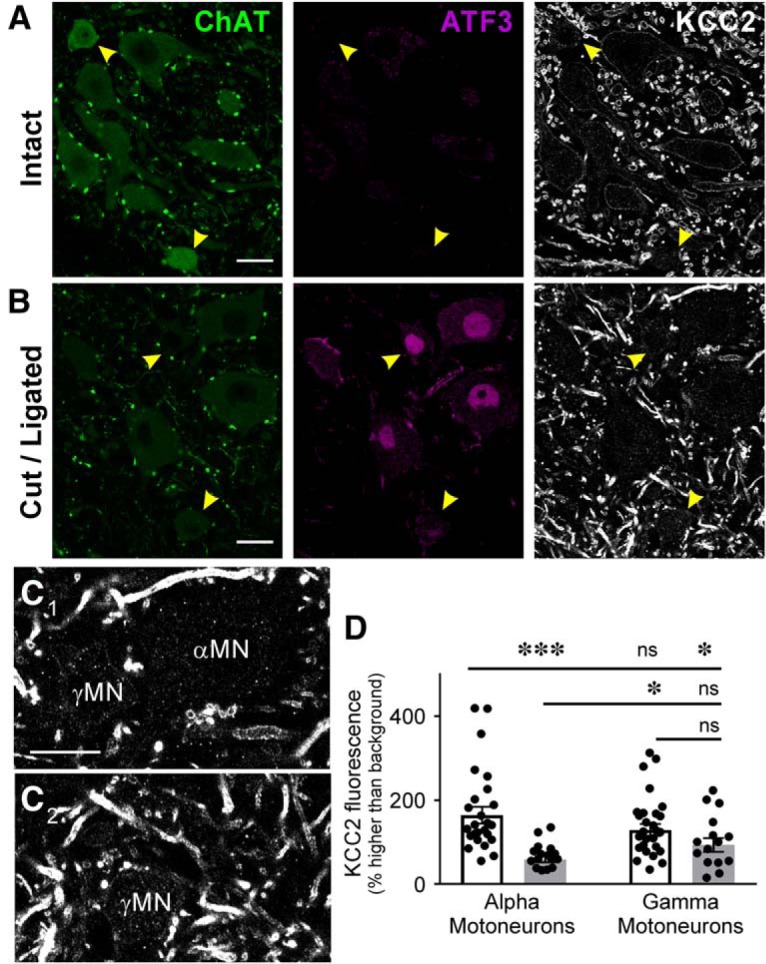
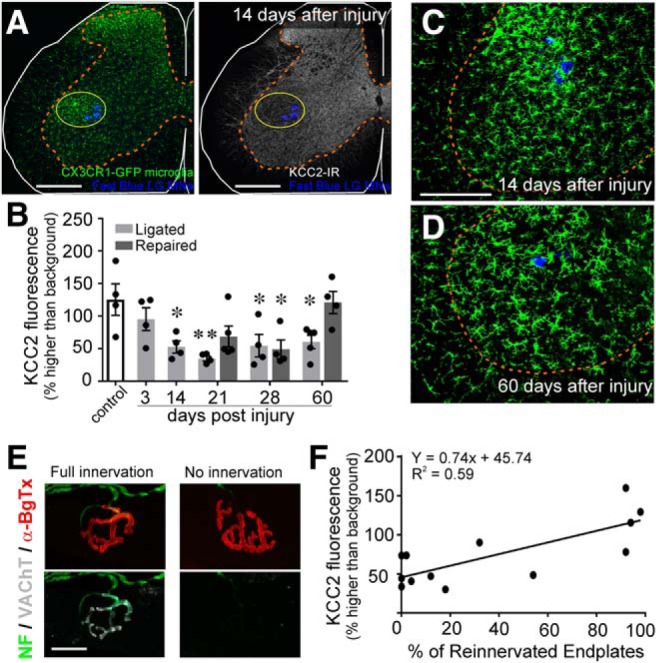
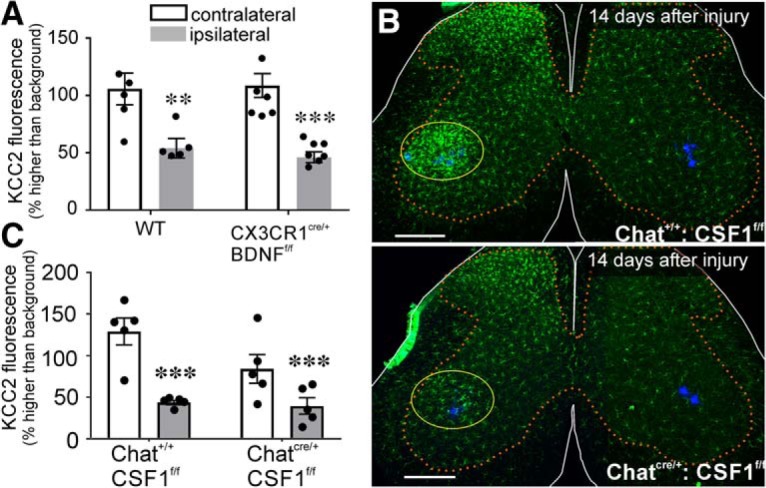
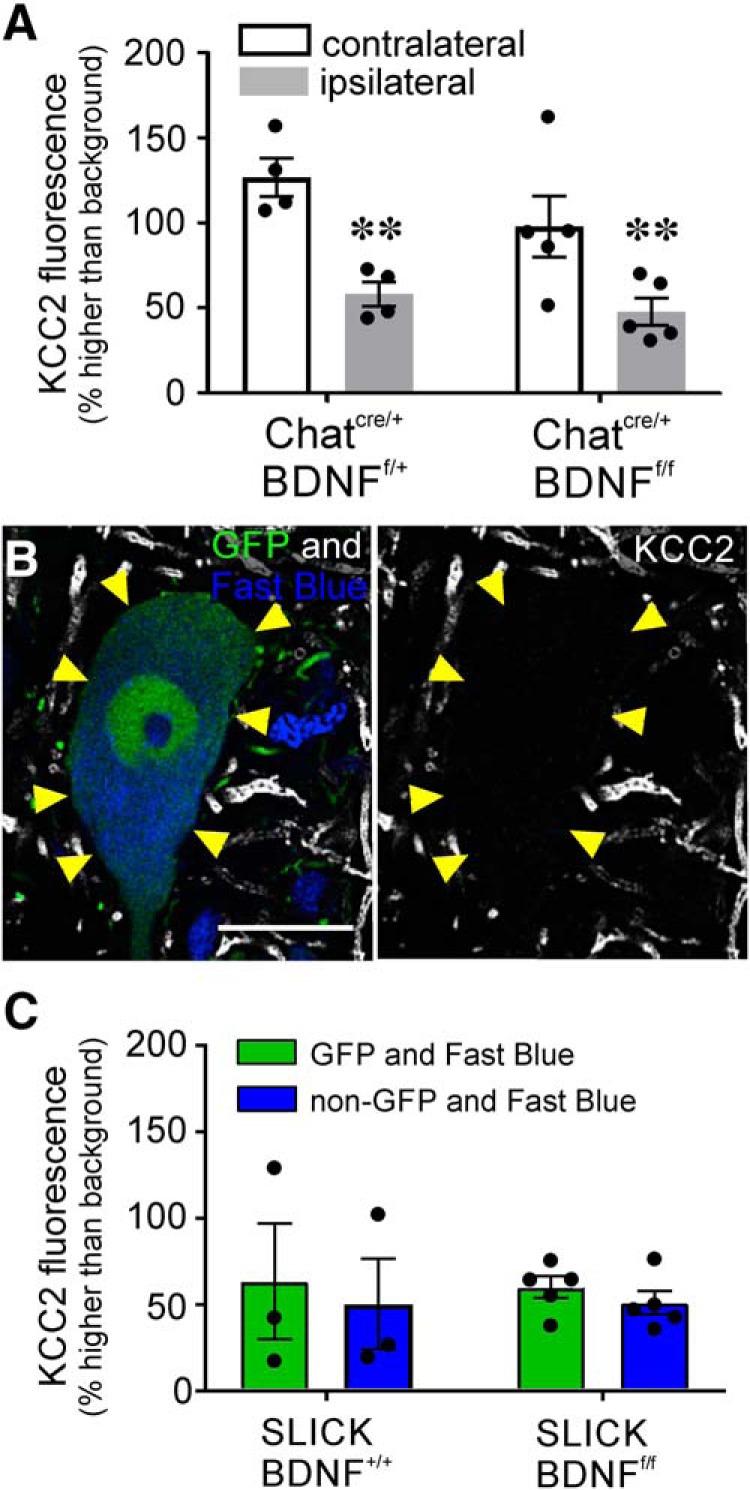
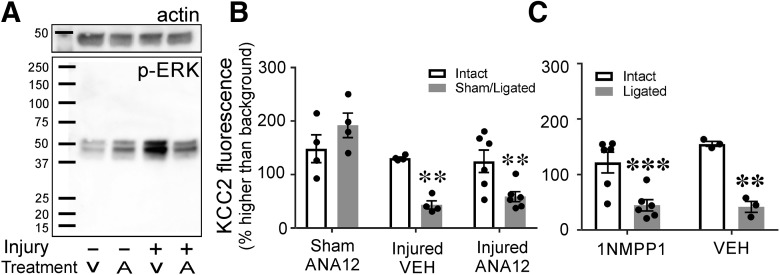
The potassium-chloride cotransporter (KCC2) maintains the low intracellular chloride found in mature central neurons and controls the strength and direction of GABA/glycine synapses. We found that following axotomy as a consequence of peripheral nerve injuries (PNIs), KCC2 protein is lost throughout the somatodendritic membrane of axotomized spinal cord motoneurons after downregulation of kcc2 mRNA expression. This large loss likely depolarizes the reversal potential of GABA/glycine synapses, resulting in GABAergic-driven spontaneous activity in spinal motoneurons similar to previous reports in brainstem motoneurons. We hypothesized that the mechanism inducing KCC2 downregulation in spinal motoneurons following peripheral axotomy might be mediated by microglia or motoneuron release of BDNF and TrkB activation as has been reported on spinal cord dorsal horn neurons after nerve injury, motoneurons after spinal cord injury (SCI), and in many other central neurons throughout development or a variety of pathologies. To test this hypothesis, we used genetic approaches to interfere with microglia activation or delete bdnf from specifically microglia or motoneurons, as well as pharmacology (ANA-12) and pharmacogenetics (F616A mice) to block TrkB activation. We show that KCC2 dysregulation in axotomized motoneurons is independent of microglia, BDNF, and TrkB. KCC2 is instead dependent on neuromuscular innervation; KCC2 levels are restored only when motoneurons reinnervate muscle. Thus, downregulation of KCC2 occurs specifically while injured motoneurons are regenerating and might be controlled by target-derived signals. GABAergic and glycinergic synapses might therefore depolarize motoneurons disconnected from their targets and contribute to augment motoneuron activity known to promote motor axon regeneration.
Keywords: GABA; KCC2; excitability; nerve injury; neuromuscular junction; regeneration.
Copyright © 2019 Akhter et al.
Figures
References
-
- Aldskogius H (2011) Mechanisms and consequences of microglial responses to peripheral axotomy. Front Biosci 3:857–868. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases