

Structure of full-length wild-type human phenylalanine hydroxylase by small angle X-ray scattering reveals substrate-induced conformational stability
- PMID: 31541188
- PMCID: PMC6754429
- DOI: 10.1038/s41598-019-49944-x
Structure of full-length wild-type human phenylalanine hydroxylase by small angle X-ray scattering reveals substrate-induced conformational stability
Erratum in
-
Author Correction: Structure of full-length wild-type human phenylalanine hydroxylase by small angle X-ray scattering reveals substrate-induced conformational stability.Sci Rep. 2019 Nov 20;9(1):17513. doi: 10.1038/s41598-019-54425-2. Sci Rep. 2019. PMID: 31745241 Free PMC article.
Abstract
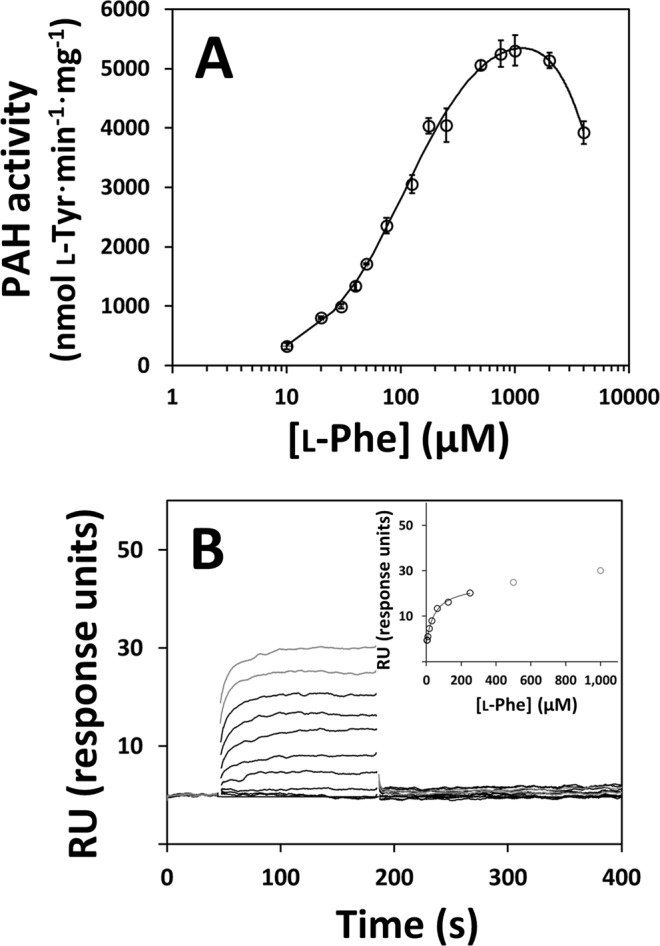
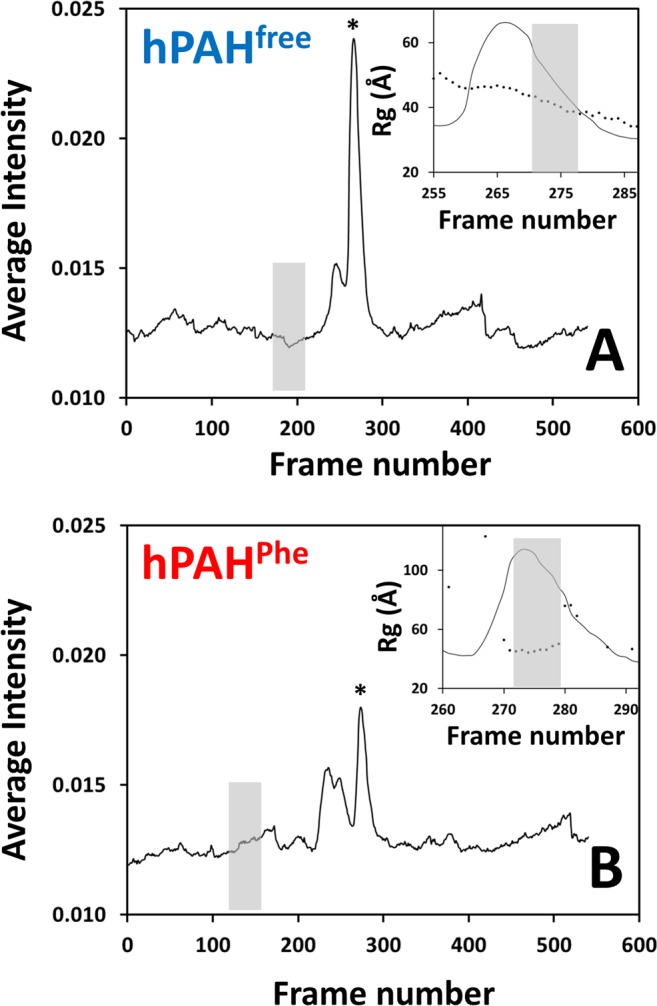
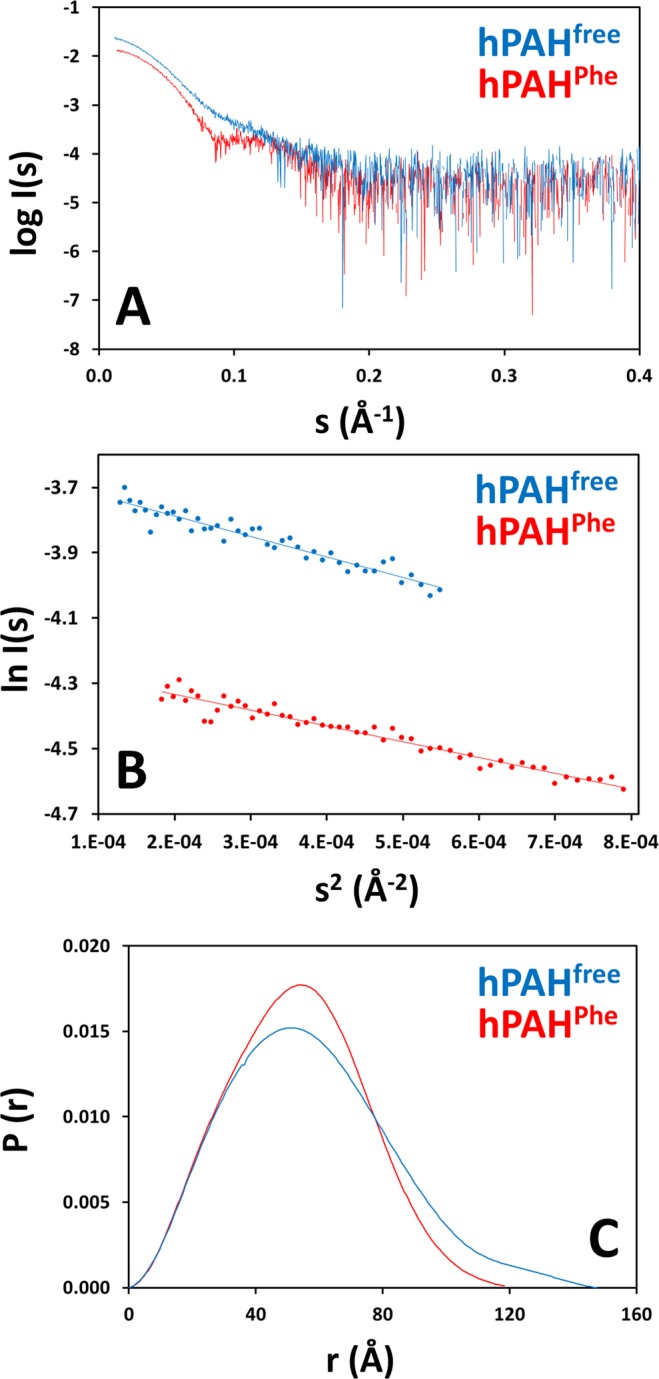
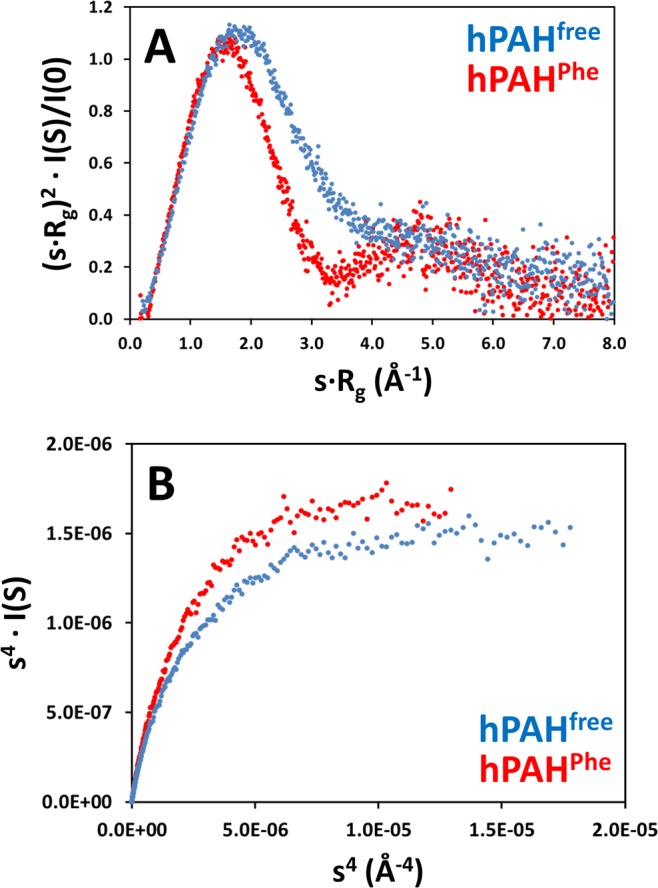
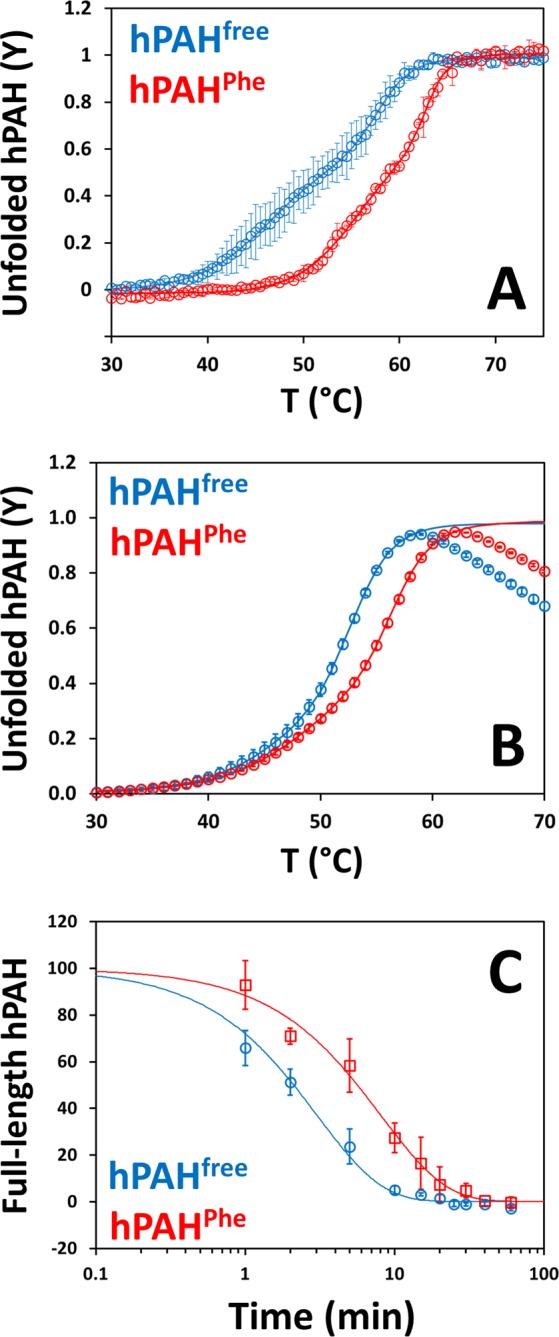
Human phenylalanine hydroxylase (hPAH) hydroxylates L-phenylalanine (L-Phe) to L-tyrosine, a precursor for neurotransmitter biosynthesis. Phenylketonuria (PKU), caused by mutations in PAH that impair PAH function, leads to neurological impairment when untreated. Understanding the hPAH structural and regulatory properties is essential to outline PKU pathophysiological mechanisms. Each hPAH monomer comprises an N-terminal regulatory, a central catalytic and a C-terminal oligomerisation domain. To maintain physiological L-Phe levels, hPAH employs complex regulatory mechanisms. Resting PAH adopts an auto-inhibited conformation where regulatory domains block access to the active site. L-Phe-mediated allosteric activation induces a repositioning of the regulatory domains. Since a structure of activated wild-type hPAH is lacking, we addressed hPAH L-Phe-mediated conformational changes and report the first solution structure of the allosterically activated state. Our solution structures obtained by small-angle X-ray scattering support a tetramer with distorted P222 symmetry, where catalytic and oligomerisation domains form a core from which regulatory domains protrude, positioning themselves close to the active site entrance in the absence of L-Phe. Binding of L-Phe induces a large movement and dimerisation of regulatory domains, exposing the active site. Activated hPAH is more resistant to proteolytic cleavage and thermal denaturation, suggesting that the association of regulatory domains stabilises hPAH.
Conflict of interest statement
The authors declare no competing interests.
Figures


References
-
- Erlandsen H, Patch MG, Gamez A, Straub M, Stevens RC. Structural studies on phenylalanine hydroxylase and implications toward understanding and treating phenylketonuria. Pediatrics. 2003;112(6 Pt 2):1557–65. - PubMed
-
- Andersen OA, Flatmark T, Hough E. High resolution crystal structures of the catalytic domain of human phenylalanine hydroxylase in its catalytically active Fe(II) form and binary complex with tetrahydrobiopterin. J Mol Biol. 2001;314(2):279–91. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
