

A case of female Fabry disease revealed by renal biopsy
- PMID: 31542871
- PMCID: PMC6990169
- DOI: 10.1007/s13730-019-00420-5
A case of female Fabry disease revealed by renal biopsy
Abstract
Fabry disease (FD) is an X-linked inherited glycosphingolipid metabolism disorder, therefore, heterozygous female FD patients display highly variable clinical symptoms, disease severity, and pathological findings. This makes it very challenging to diagnosing female patients with FD. A 69-year-old Japanese female was introduced to the nephrologist for the evaluation of proteinuria. A renal biopsy was performed. Although the light microscopic examinations revealed that most of the glomeruli showed minor glomerular abnormalities, however, vacuolation was apparently found in the tubular epithelial cells. Immunofluorescence staining for globotriaosylceramide was positively detected in some podocytes and distal tubular epithelial cells. In addition, myelin-like structure (zebra body) was detected by electron microscopy. Pathological findings were most consistent with FD. Consequently, biochemical and genetic analysis confirmed the diagnosis of female FD. Enzyme replacement therapy was performed in conjunction with renin-angiotensin aldosterone system inhibitors and beta-blockers. The patient's family members received the analysis, and the same DNA missense mutation was detected in the patient's grandson. The enzyme replacement therapy was introduced to the grandson. The present case showed that renal biopsy can contribute towards a correct diagnosis for FD. Particularly, in female FD patients, careful examination of pathological changes is essential, for example, vacuolation of any type of renal cells may be a clue for the diagnosis.
Keywords: Enzyme replacement therapy; Fabry disease; Pathology; Renal biopsy; Vacuolation.
Conflict of interest statement
The authors have no competing interests to declare.
Figures


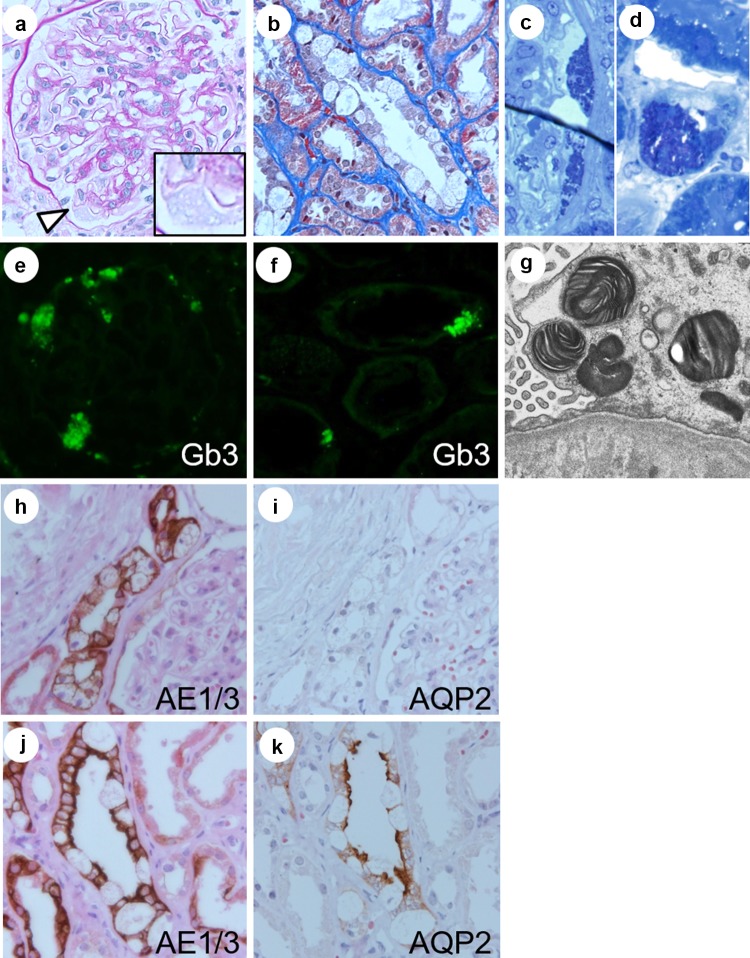
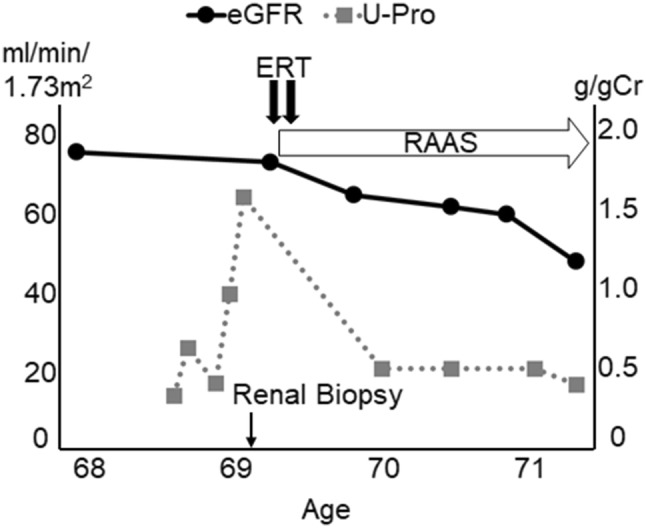
References
-
- Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L. Enzymatic defect in Fabry's disease: ceramidetrihexosidase deficiency. N Engl J Med. 1967;27:163–167. - PubMed
-
- Wilcox WR, Oliveira JP, Hopkin RJ, Ortiz A, Banikazemi M, Feldt-Rasmussen U, Sims K, Waldek S, Pastores GM, Lee P, Eng CM, Marodi L, Stanford KE, Breunig F, Wanner C, Warnock DG, Lemay RM, Germain DP. Females with Fabry disease frequently have major organ involvement: lessons from the Fabry Registry. Mol Genet Metab. 2008;93:112–128. doi: 10.1016/j.ymgme.2007.09.013. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
