

Combining the Allosteric Inhibitor Asciminib with Ponatinib Suppresses Emergence of and Restores Efficacy against Highly Resistant BCR-ABL1 Mutants
- PMID: 31543464
- PMCID: PMC6893878
- DOI: 10.1016/j.ccell.2019.08.004
Combining the Allosteric Inhibitor Asciminib with Ponatinib Suppresses Emergence of and Restores Efficacy against Highly Resistant BCR-ABL1 Mutants
Abstract
BCR-ABL1 point mutation-mediated resistance to tyrosine kinase inhibitor (TKI) therapy in Philadelphia chromosome-positive (Ph+) leukemia is effectively managed with several approved drugs, including ponatinib for BCR-ABL1T315I-mutant disease. However, therapy options are limited for patients with leukemic clones bearing multiple BCR-ABL1 mutations. Asciminib, an allosteric inhibitor targeting the myristoyl-binding pocket of BCR-ABL1, is active against most single mutants but ineffective against all tested compound mutants. We demonstrate that combining asciminib with ATP site TKIs enhances target inhibition and suppression of resistant outgrowth in Ph+ clinical isolates and cell lines. Inclusion of asciminib restores ponatinib's effectiveness against currently untreatable compound mutants at clinically achievable concentrations. Our findings support combining asciminib with ponatinib as a treatment strategy for this molecularly defined group of patients.
Keywords: ABL001; allosteric inhibitors; asciminib; chronic myeloid leukemia; compound mutation; ponatinib; targeted therapy.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
Disclosure of Potential Conflicts of Interest
M.W.D. served on advisory boards and as a consultant for Bristol-Myers Squibb, ARIAD, and Novartis and receives research funding from Bristol-Myers Squibb, Celgene, Novartis and Gilead. B.J.D. potential competing interests -- SAB: Aileron Therapeutics, ALLCRON, Cepheid, Gilead Sciences, Vivid Biosciences, Celgene & Baxalta (inactive); SAB & Stock: Aptose Biosciences, Blueprint Medicines, Beta Cat, GRAIL, Third Coast Therapeutics, CTI BioPharma (inactive); Scientific Founder & Stock: MolecularMD; Board of Directors & Stock: Amgen; Board of Directors: Burroughs Wellcome Fund, CureOne; Joint Steering Committee: Beat AML LLS; Clinical Trial Funding: Novartis, Bristol-Myers Squibb, Pfizer; Royalties from Patent 6958335 (Novartis exclusive license) and OHSU and Dana-Farber Cancer Institute (one Merck exclusive license).
Figures
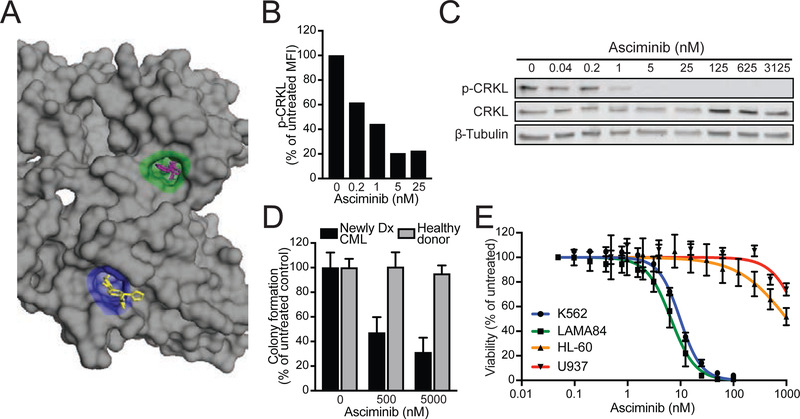
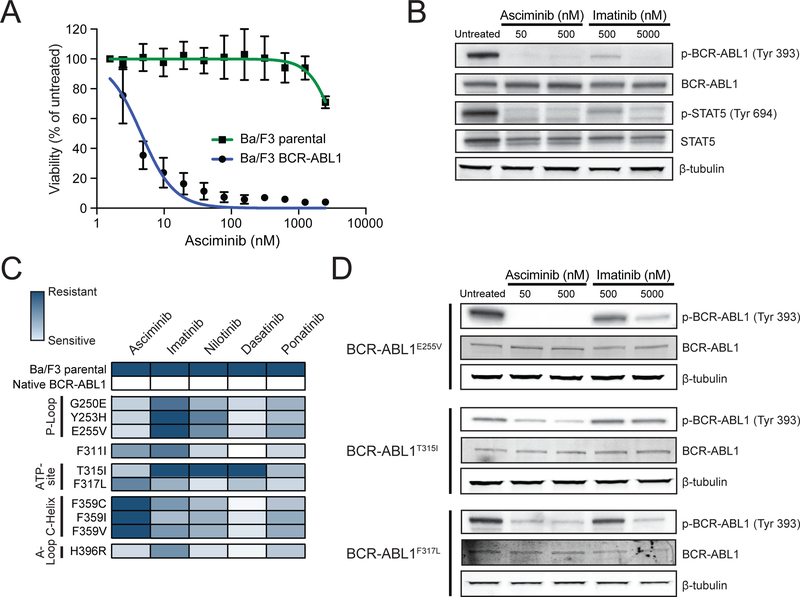
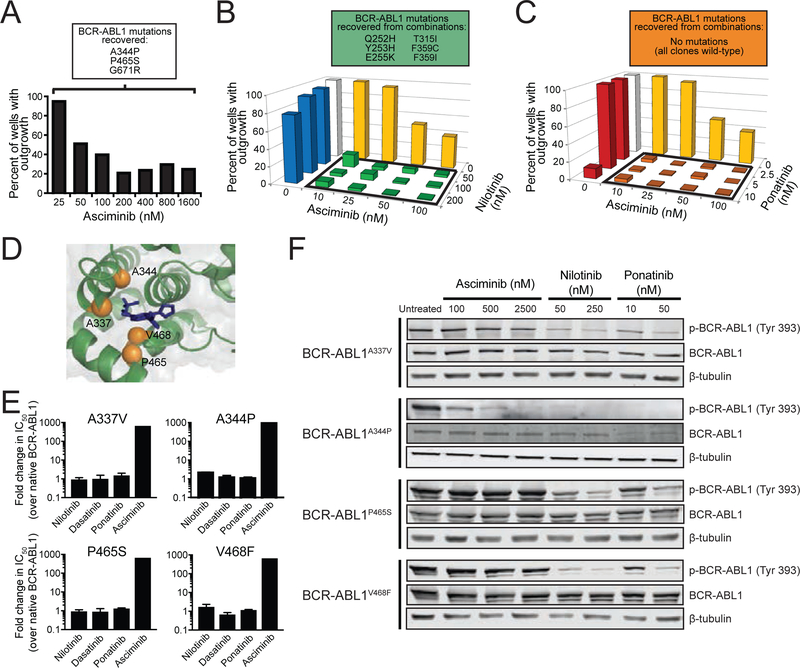
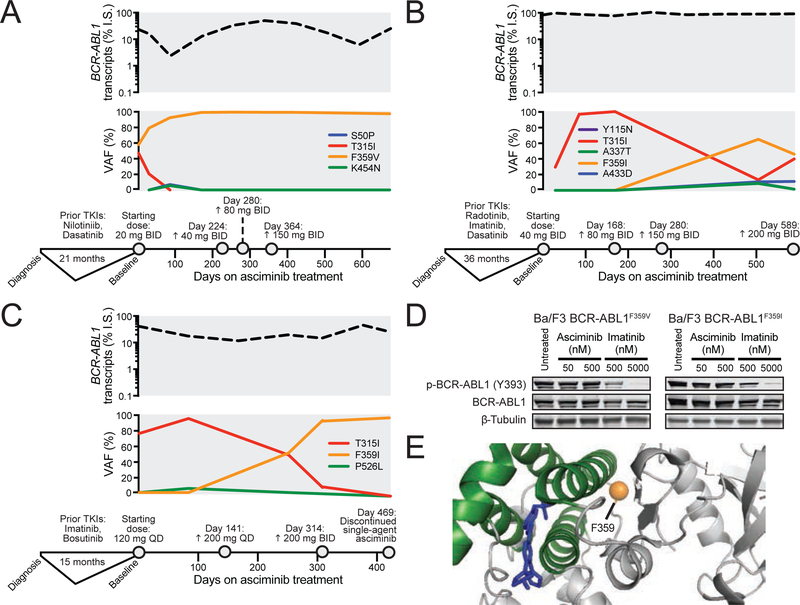
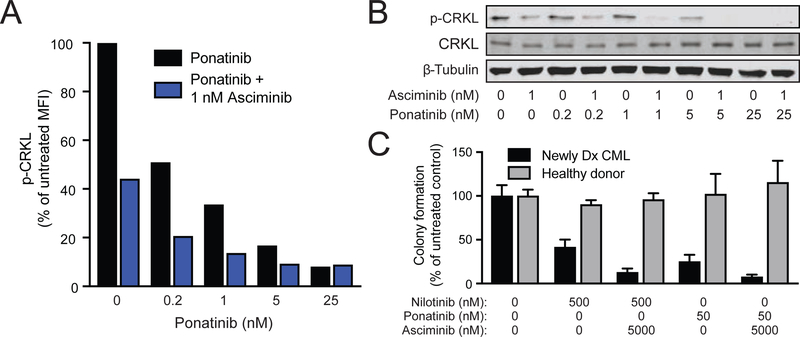
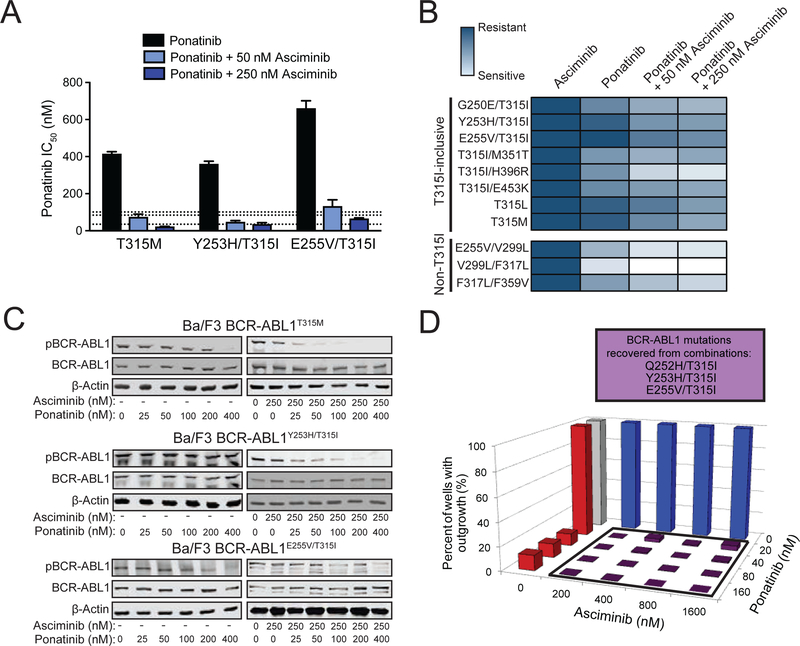
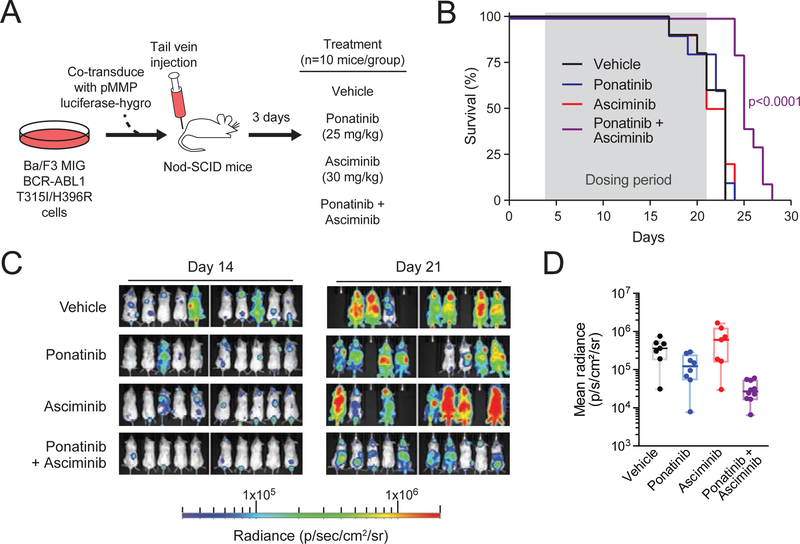
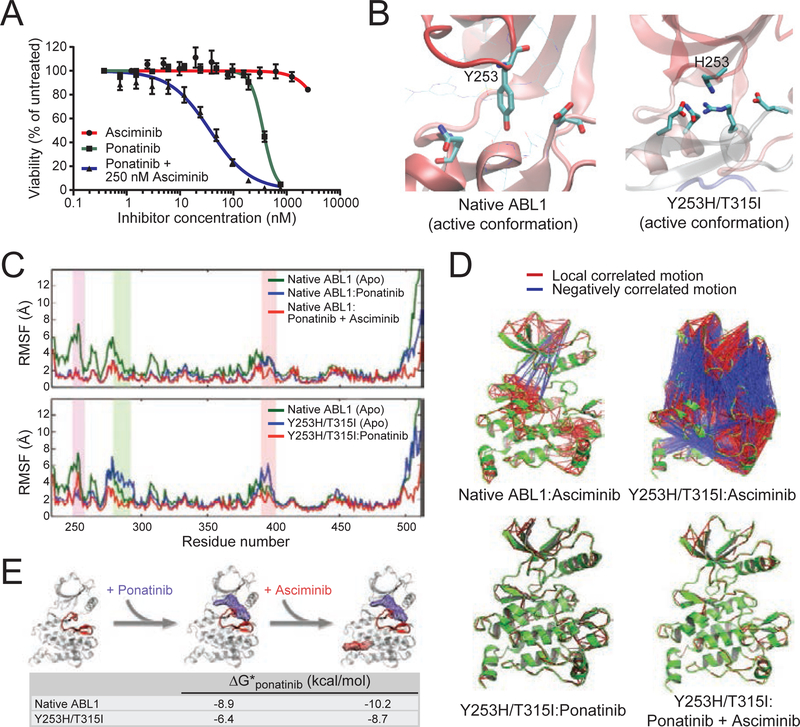
References
-
- Bradeen HA, Eide CA, O’Hare T, Johnson KJ, Willis SG, Lee FY, Druker BJ, and Deininger MW (2006). Comparison of imatinib, dasatinib (BMS-354825), and nilotinib (AMN107) in an n-ethyl-n-nitrosourea (ENU)-based mutagenesis screen: high efficacy of drug combinations. Blood 108, 2332–2338. - PMC - PubMed
-
- Breccia M, Pregno P, Spallarossa P, Arboscello E, Ciceri F, Giorgi M, Grossi A, Mallardo M, Nodari S, Ottolini S, et al. (2017). Identification, prevention and management of cardiovascular risk in chronic myeloid leukaemia patients candidate to ponatinib: an expert opinion. Annals of hematology 96, 549–558. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
