

Bispecific T-Cell Redirection versus Chimeric Antigen Receptor (CAR)-T Cells as Approaches to Kill Cancer Cells
- PMID: 31544847
- PMCID: PMC6784091
- DOI: 10.3390/antib8030041
Bispecific T-Cell Redirection versus Chimeric Antigen Receptor (CAR)-T Cells as Approaches to Kill Cancer Cells
Abstract
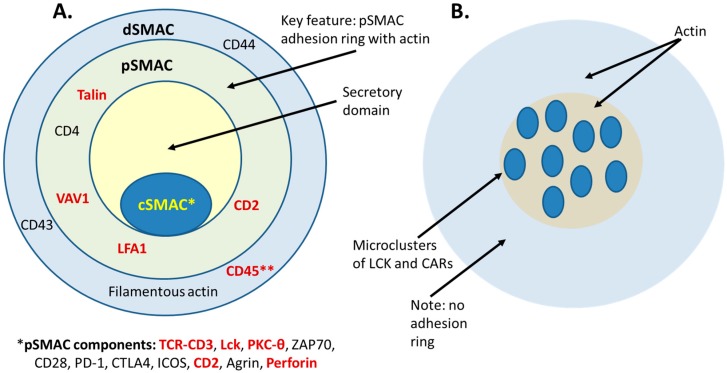
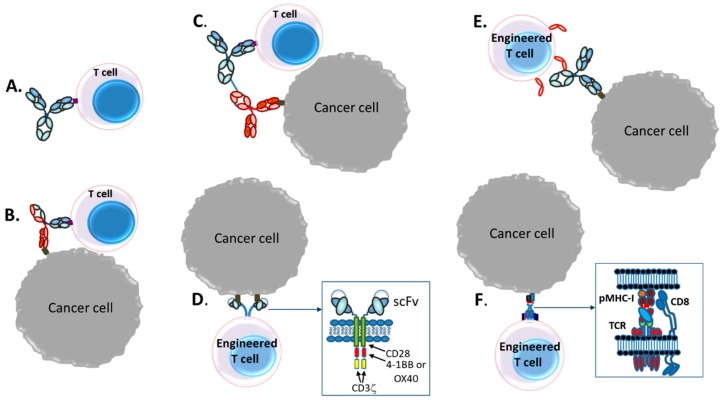
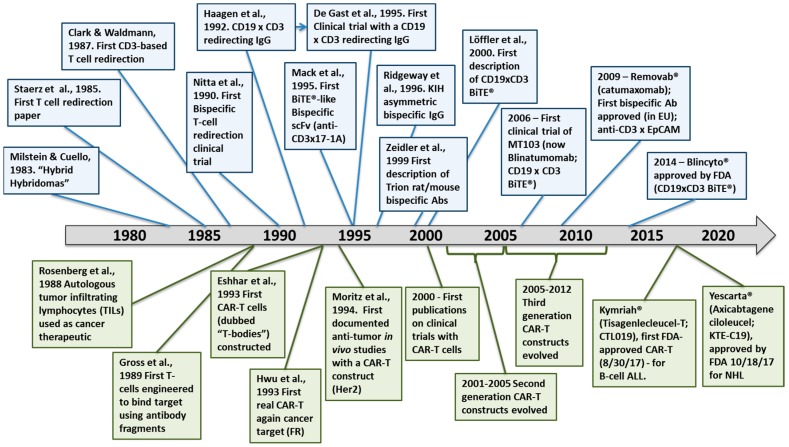
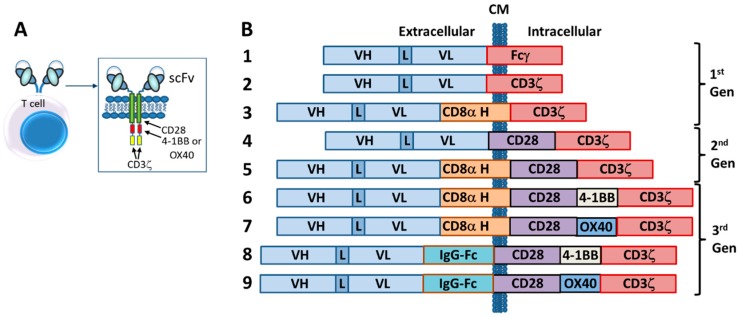
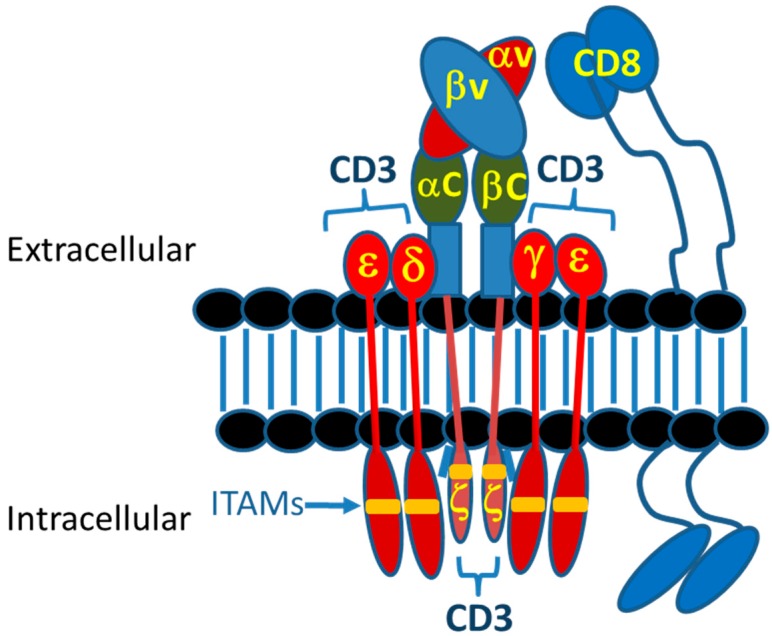
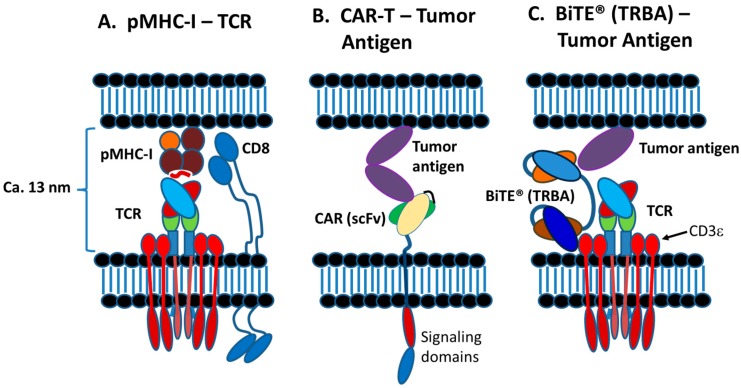
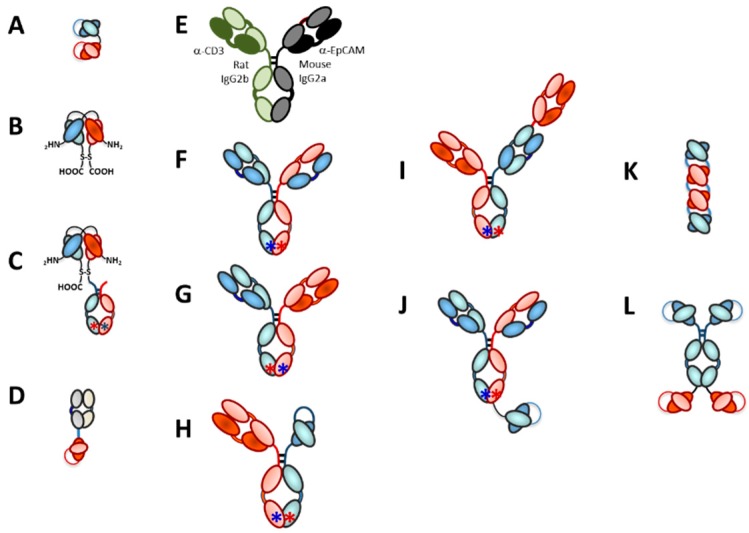
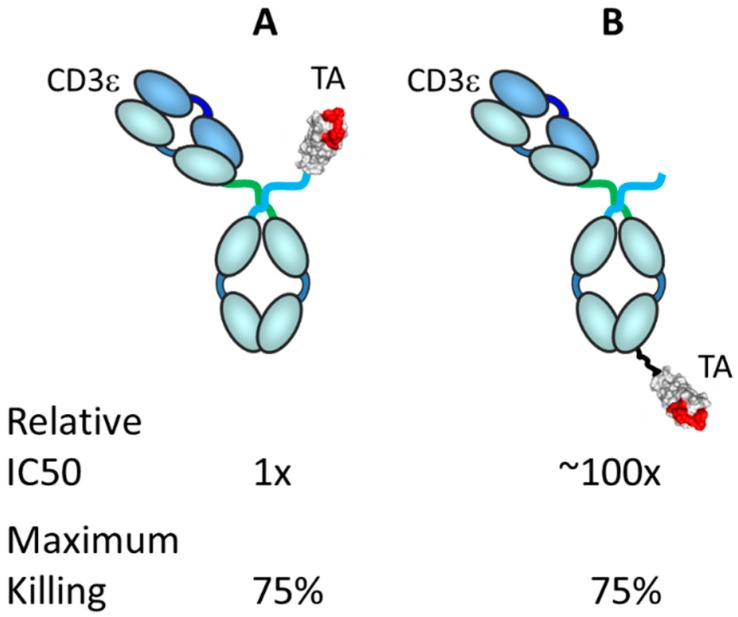
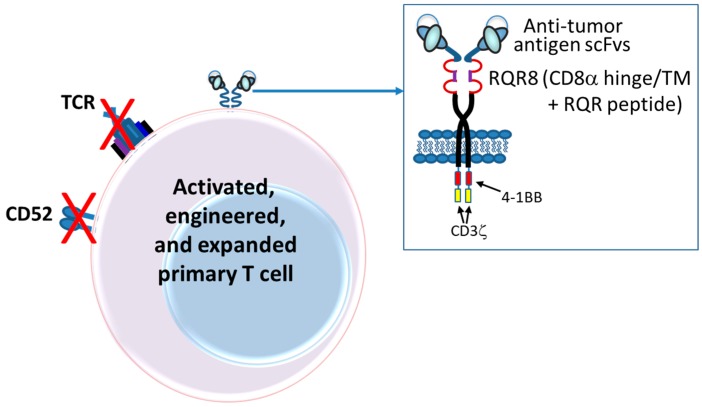
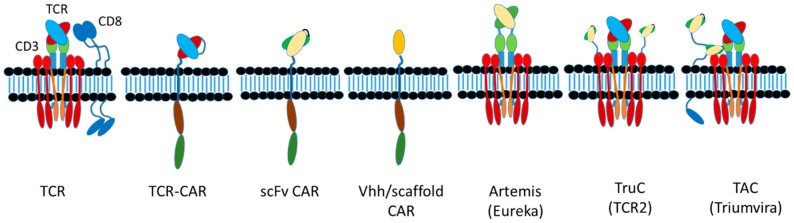
The concepts for T-cell redirecting bispecific antibodies (TRBAs) and chimeric antigen receptor (CAR)-T cells are both at least 30 years old but both platforms are just now coming into age. Two TRBAs and two CAR-T cell products have been approved by major regulatory agencies within the last ten years for the treatment of hematological cancers and an additional 53 TRBAs and 246 CAR cell constructs are in clinical trials today. Two major groups of TRBAs include small, short-half-life bispecific antibodies that include bispecific T-cell engagers (BiTE®s) which require continuous dosing and larger, mostly IgG-like bispecific antibodies with extended pharmacokinetics that can be dosed infrequently. Most CAR-T cells today are autologous, although significant strides are being made to develop off-the-shelf, allogeneic CAR-based products. CAR-Ts form a cytolytic synapse with target cells that is very different from the classical immune synapse both physically and mechanistically, whereas the TRBA-induced synapse is similar to the classic immune synapse. Both TRBAs and CAR-T cells are highly efficacious in clinical trials but both also present safety concerns, particularly with cytokine release syndrome and neurotoxicity. New formats and dosing paradigms for TRBAs and CAR-T cells are being developed in efforts to maximize efficacy and minimize toxicity, as well as to optimize use with both solid and hematologic tumors, both of which present significant challenges such as target heterogeneity and the immunosuppressive tumor microenvironment.
Keywords: CD3ε, T cells; NK cells; T-cell redirection; bispecific antibody; chimeric antigen receptor; immune synapse; tumor cell killing; tumor microenvironment.
Conflict of interest statement
The authors declare the following potential conflicts of interest: W.R.S. and M.N. both were recent employees at Janssen R&D, Johnson & Johnson and hold stock in that company. W.R.S. currently is a consultant for several small biotechnology companies and is a member of the Board of Directors of IGM Biosciences. M.N. currently is an employee of Century Therapeutics, Philadelphia, PA.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
