Serial Anti-Myelin Oligodendrocyte Glycoprotein Antibody Analyses and Outcomes in Children With Demyelinating Syndromes
- PMID: 31545352
- PMCID: PMC6763982
- DOI: 10.1001/jamaneurol.2019.2940
Serial Anti-Myelin Oligodendrocyte Glycoprotein Antibody Analyses and Outcomes in Children With Demyelinating Syndromes
Abstract
Importance: Identifying the course of demyelinating disease associated with myelin oligodendrocyte glycoprotein (MOG) autoantibodies is critical to guide appropriate treatment choices.
Objective: To characterize serial anti-MOG antibody serologies and clinical and imaging features at presentation and during follow-up in an inception cohort of prospectively monitored children with acquired demyelination.
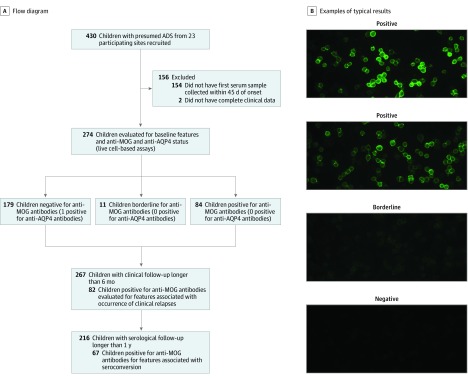
Design, setting, and participants: In this prospective cohort study, study participants were recruited from July 2004 to February 2017 through the multicenter Canadian Pediatric Demyelinating Disease Study. Inclusion criteria included (1) incident central nervous system demyelination, (2) at least 1 serum sample obtained within 45 days from onset, and (3) complete clinical information. Of 430 participants with acquired demyelinating syndrome recruited, 274 were included in analyses. Of 156 excluded participants, 154 were excluded owing to missing baseline samples and 2 owing to incomplete clinical information. Data were analyzed from May to October 2018.
Main outcomes and measures: Presence of anti-MOG antibodies was blindly assessed in serial samples collected over a median of 4 years. Clinical, magnetic resonance imaging, and cerebrospinal fluid features were characterized at presentation, and subsequent disease course was assessed by development of new brain magnetic resonance imaging lesions, total lesion volume at last evaluation, annualized relapse rates, Expanded Disability Status Scale score and visual functional score at 4 years, and any disease-modifying treatment exposure.
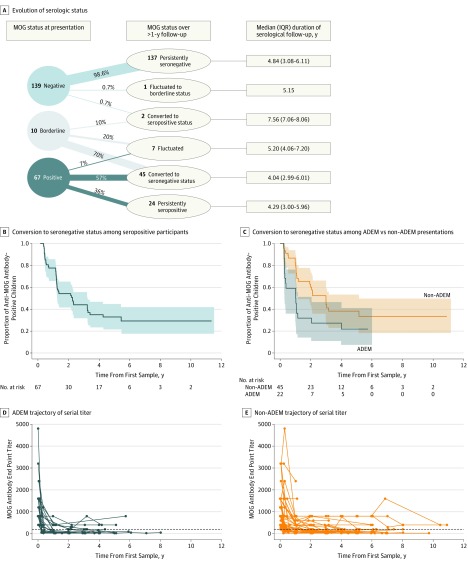
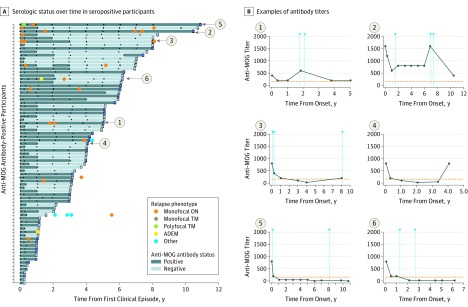
Results: Of the 274 included participants, 140 (51.1%) were female, and the median (interquartile range) age of all participants was 10.8 (6.2-13.9) years. One-third of children were positive for anti-MOG antibodies at the time of incident demyelination. Clinical presentations included a combination of optic neuritis, transverse myelitis, and acute disseminated encephalomyelitis for 81 of 84 anti-MOG antibody-positive children (96%). Brain lesions were present in 51 of 76 anti-MOG antibody-positive participants (67%), but magnetic resonance imaging characteristics differed with age at presentation. Complete resolution of baseline lesions was observed in 26 of 49 anti-MOG antibody-positive participants (53%). On serial serum analysis, 38 of 67 participants (57%) who were seropositive at onset became seronegative (median time to conversion, 1 year). Among all participants who were positive for anti-MOG antibodies at presentation, clinical relapses occurred in 9 of 24 children (38%) who remained persistently seropositive and in 5 of 38 children (13%) who converted to seronegative status.
Conclusions and relevance: Myelin oligodendrocyte glycoprotein antibodies are common in children with acquired demyelinating syndrome and are transient in approximatively half of cases. Even when persistently positive, most anti-MOG antibody-positive children experience a monophasic disease. The presence of anti-MOG antibodies at the time of incident demyelination should not immediately prompt the initiation of long-term immunomodulatory therapy.
Conflict of interest statement
Figures