

Immunoglobulin-driven Complement Activation Regulates Proinflammatory Remodeling in Pulmonary Hypertension
- PMID: 31545648
- PMCID: PMC6961733
- DOI: 10.1164/rccm.201903-0591OC
Immunoglobulin-driven Complement Activation Regulates Proinflammatory Remodeling in Pulmonary Hypertension
Abstract
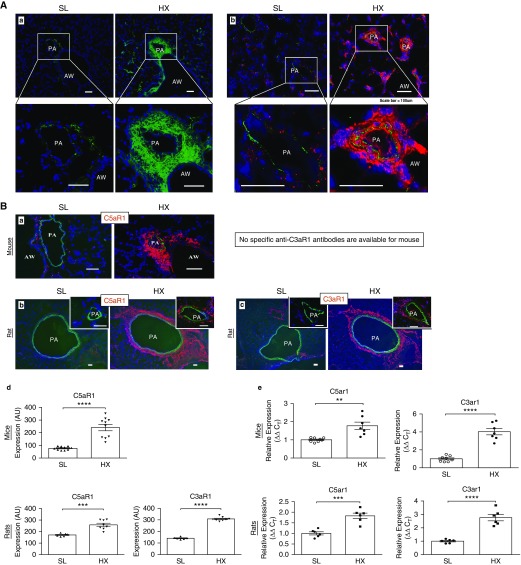
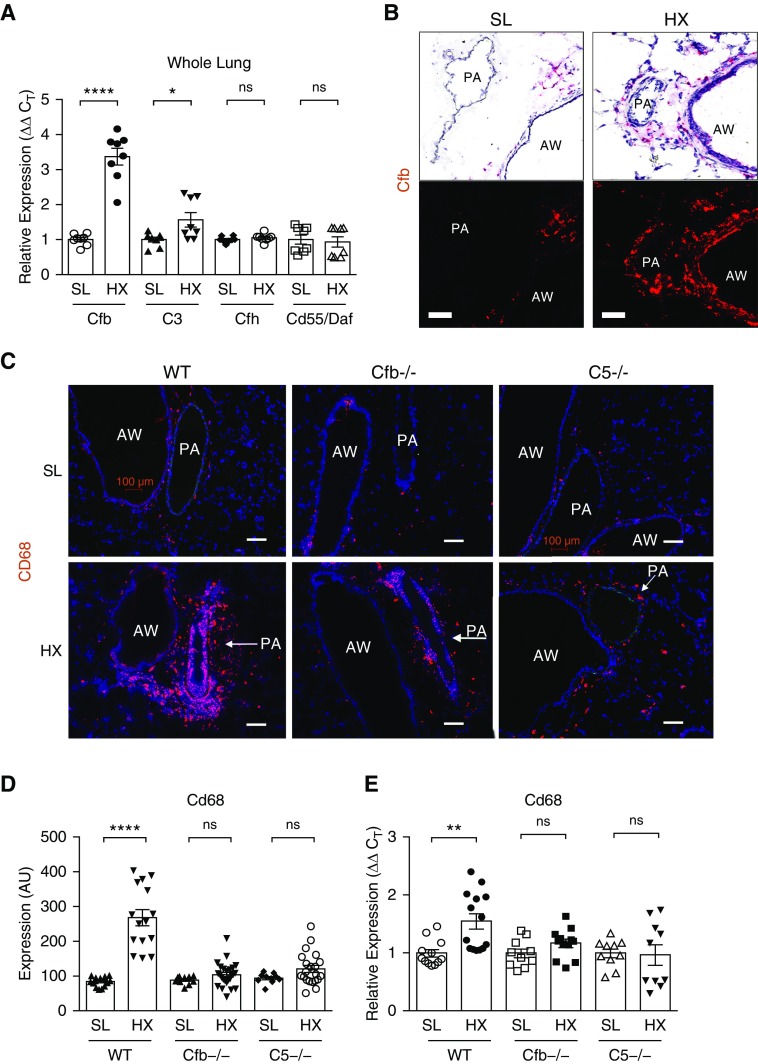
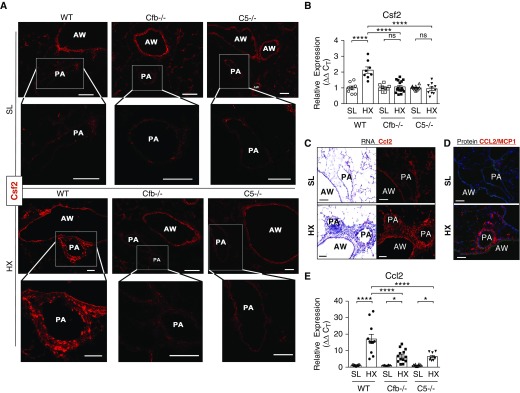
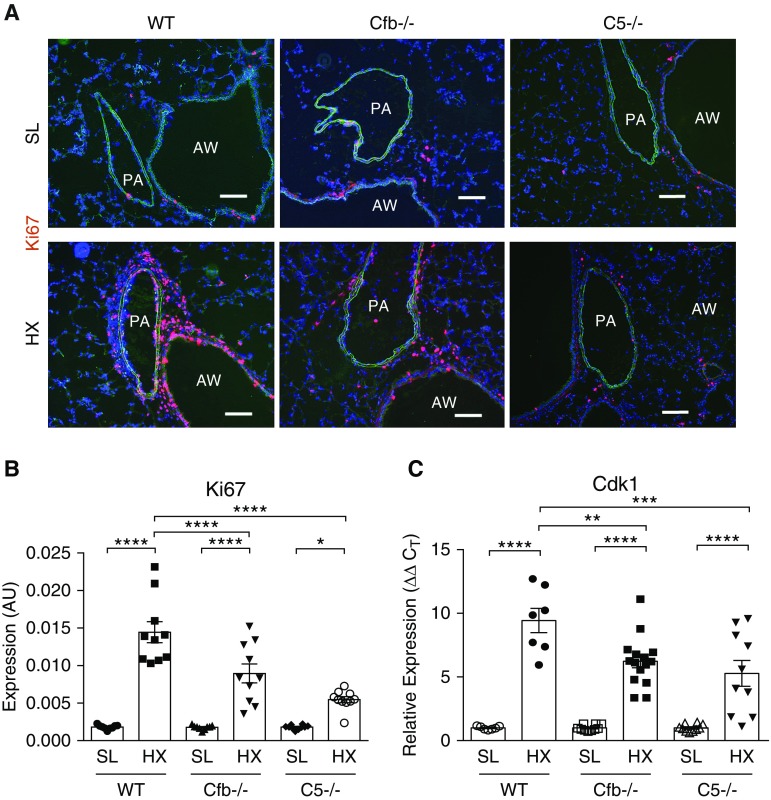
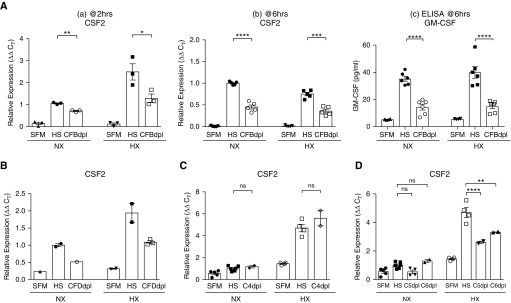
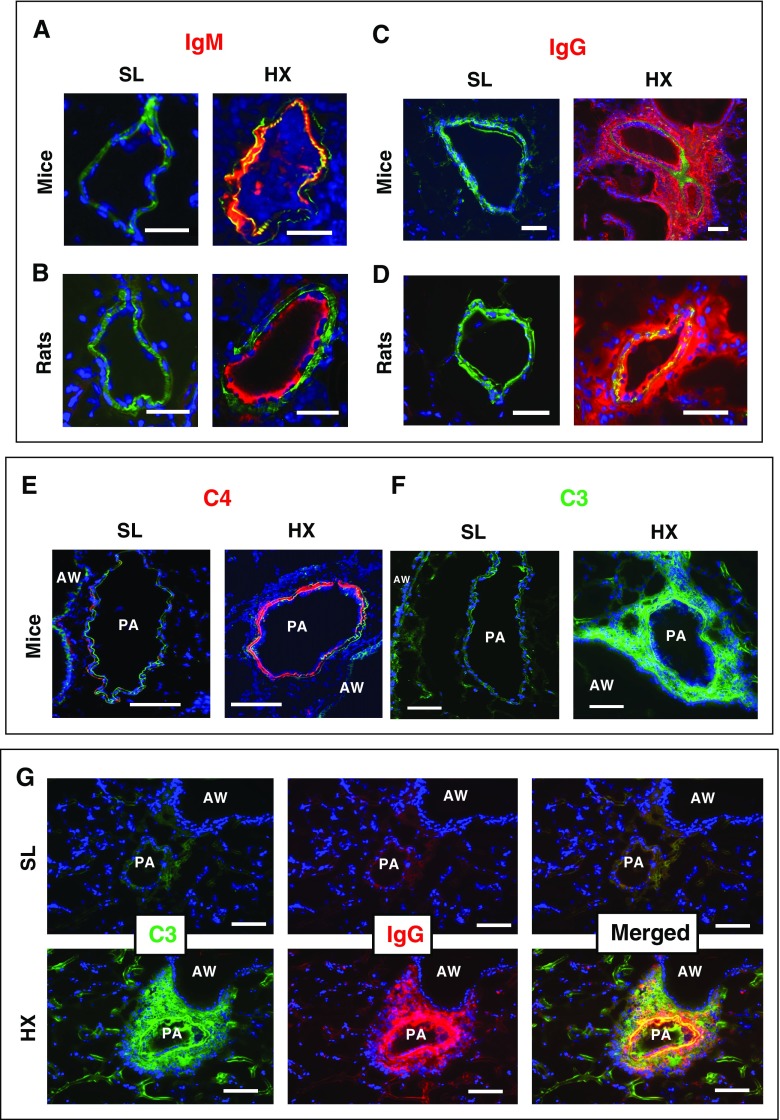
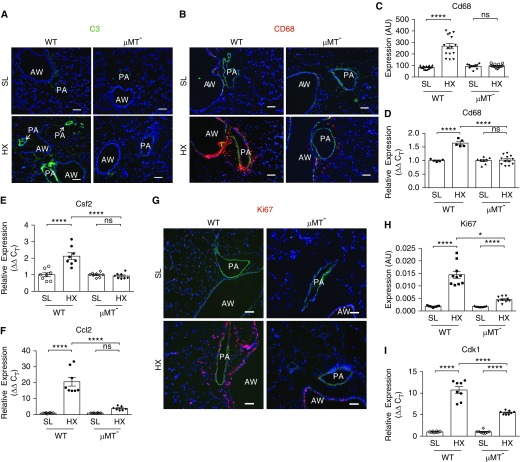
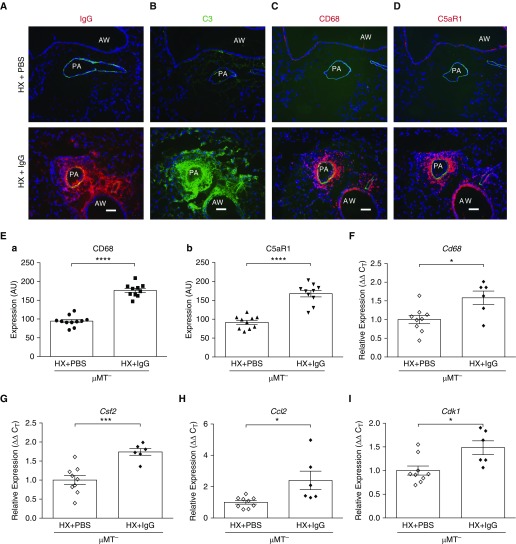
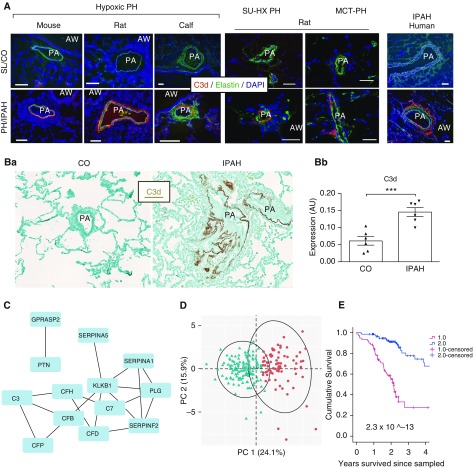
Rationale: Pulmonary hypertension (PH) is a life-threatening cardiopulmonary disorder in which inflammation and immunity have emerged as critical early pathogenic elements. Although proinflammatory processes in PH and pulmonary arterial hypertension (PAH) are the focus of extensive investigation, the initiating mechanisms remain elusive.Objectives: We tested whether activation of the complement cascade is critical in regulating proinflammatory and pro-proliferative processes in the initiation of experimental hypoxic PH and can serve as a prognostic biomarker of outcome in human PAH.Methods: We used immunostaining of lung tissues from experimental PH models and patients with PAH, analyses of genetic murine models lacking specific complement components or circulating immunoglobulins, cultured human pulmonary adventitial fibroblasts, and network medicine analysis of a biomarker risk panel from plasma of patients with PAH.Measurements and Main Results: Pulmonary perivascular-specific activation of the complement cascade was identified as a consistent critical determinant of PH and PAH in experimental animal models and humans. In experimental hypoxic PH, proinflammatory and pro-proliferative responses were dependent on complement (alternative pathway and component 5), and immunoglobulins, particularly IgG, were critical for activation of the complement cascade. We identified Csf2/GM-CSF as a primary complement-dependent inflammatory mediator. Furthermore, using network medicine analysis of a biomarker risk panel from plasma of patients with PAH, we demonstrated that complement signaling can serve as a prognostic factor for clinical outcome in PAH.Conclusions: This study establishes immunoglobulin-driven dysregulated complement activation as a critical pathobiological mechanism regulating proinflammatory and pro-proliferative processes in the initiation of experimental hypoxic PH and demonstrates complement signaling as a critical determinant of clinical outcome in PAH.
Keywords: GM-CSF; biomarkers; hypoxia; inflammation; vascular remodeling.
Figures
Comment in
-
An Emotional Molecular Pathway in Pulmonary Hypertension-Alternative Complement System.Am J Respir Crit Care Med. 2020 Jan 15;201(2):138-140. doi: 10.1164/rccm.201909-1790ED. Am J Respir Crit Care Med. 2020. PMID: 31600450 Free PMC article. No abstract available.
References
-
- Nicolls MR, Taraseviciene-Stewart L, Rai PR, Badesch DB, Voelkel NF. Autoimmunity and pulmonary hypertension: a perspective. Eur Respir J. 2005;26:1110–1118. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
