

Intragenomic variability and extended sequence patterns in the mutational signature of ultraviolet light
- PMID: 31548379
- PMCID: PMC6789905
- DOI: 10.1073/pnas.1909021116
Intragenomic variability and extended sequence patterns in the mutational signature of ultraviolet light
Abstract
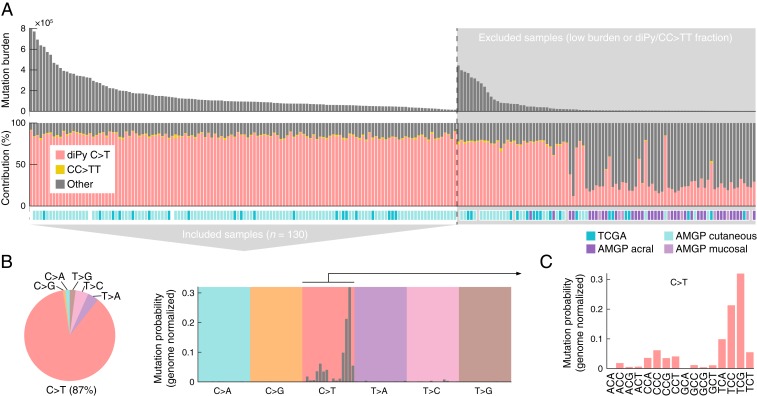
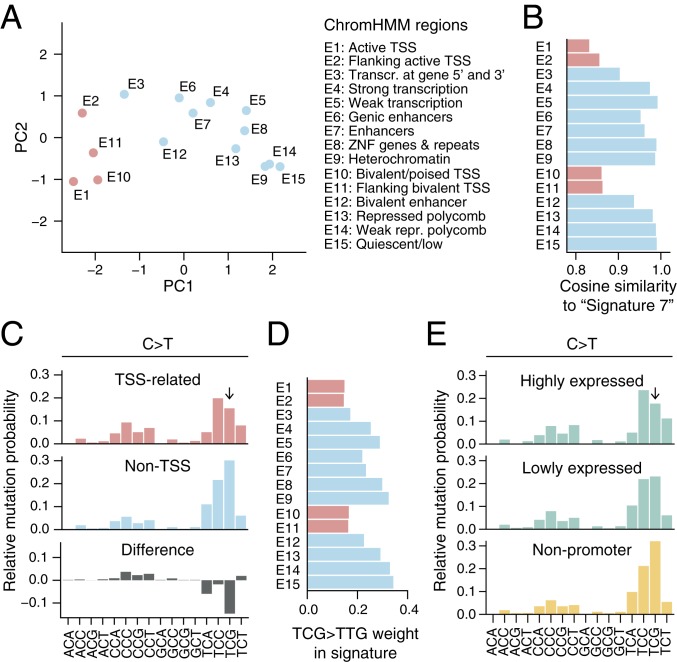
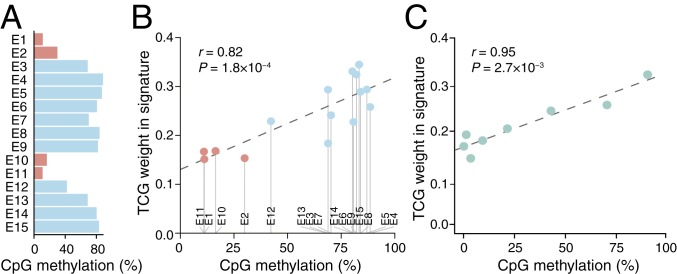
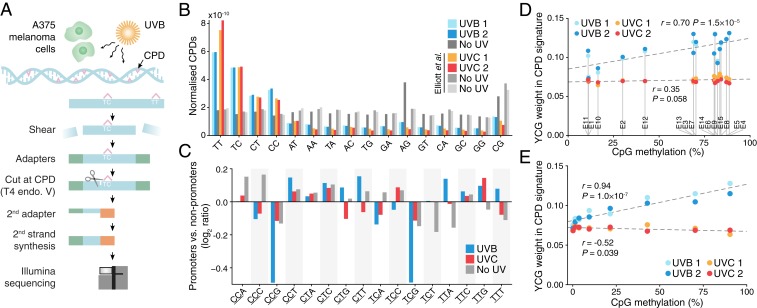
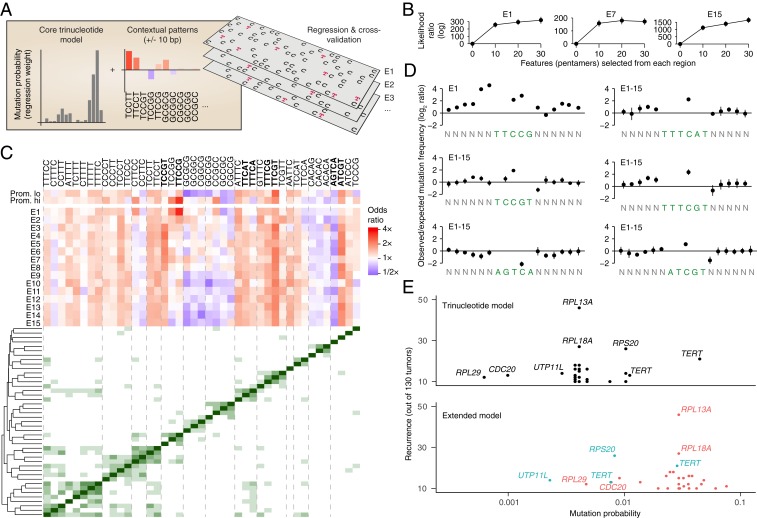
Mutational signatures can reveal properties of underlying mutational processes and are important when assessing signals of selection in cancer. Here, we describe the sequence characteristics of mutations induced by ultraviolet (UV) light, a major mutagen in several human cancers, in terms of extended (longer than trinucleotide) patterns as well as variability of the signature across chromatin states. Promoter regions display a distinct UV signature with reduced TCG > TTG transitions, and genome-wide mapping of UVB-induced DNA photoproducts (pyrimidine dimers) showed that this may be explained by decreased damage formation at hypomethylated promoter CpG sites. Further, an extended signature model encompassing additional information from longer contextual patterns improves modeling of UV mutations, which may enhance discrimination between drivers and passenger events. Our study presents a refined picture of the UV signature and underscores that the characteristics of a single mutational process may vary across the genome.
Keywords: DNA damage; UV; cancer genomics; mutational signature; pyrimidine dimer.
Copyright © 2019 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
