

SHERLOCK: nucleic acid detection with CRISPR nucleases
- PMID: 31548639
- PMCID: PMC6956564
- DOI: 10.1038/s41596-019-0210-2
SHERLOCK: nucleic acid detection with CRISPR nucleases
Erratum in
-
Author Correction: SHERLOCK: nucleic acid detection with CRISPR nucleases.Nat Protoc. 2020 Mar;15(3):1311. doi: 10.1038/s41596-020-0302-z. Nat Protoc. 2020. PMID: 32005984
Abstract
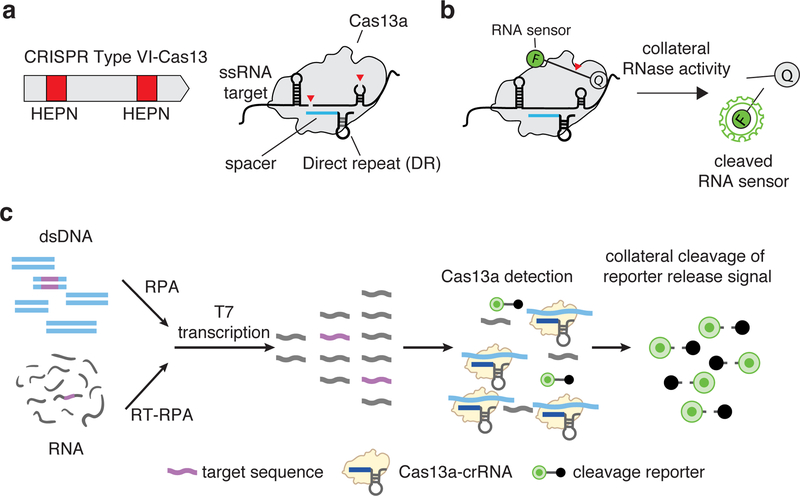
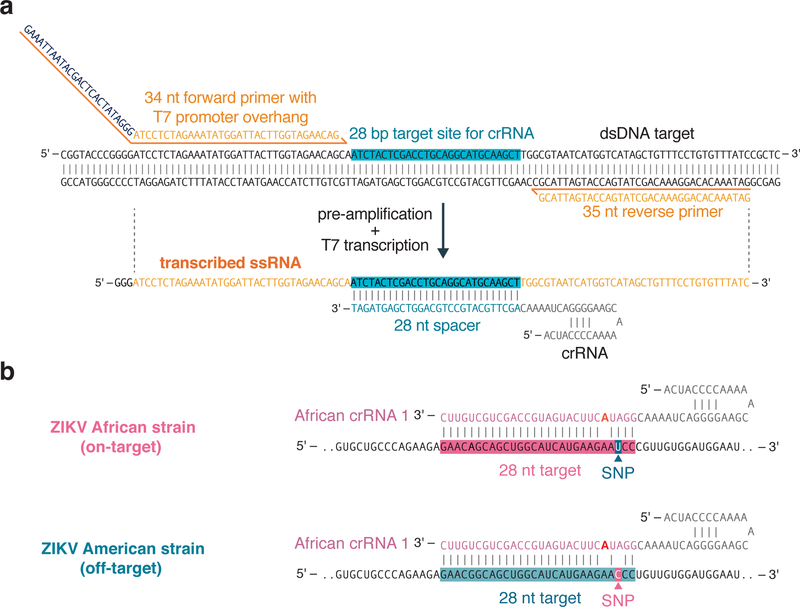
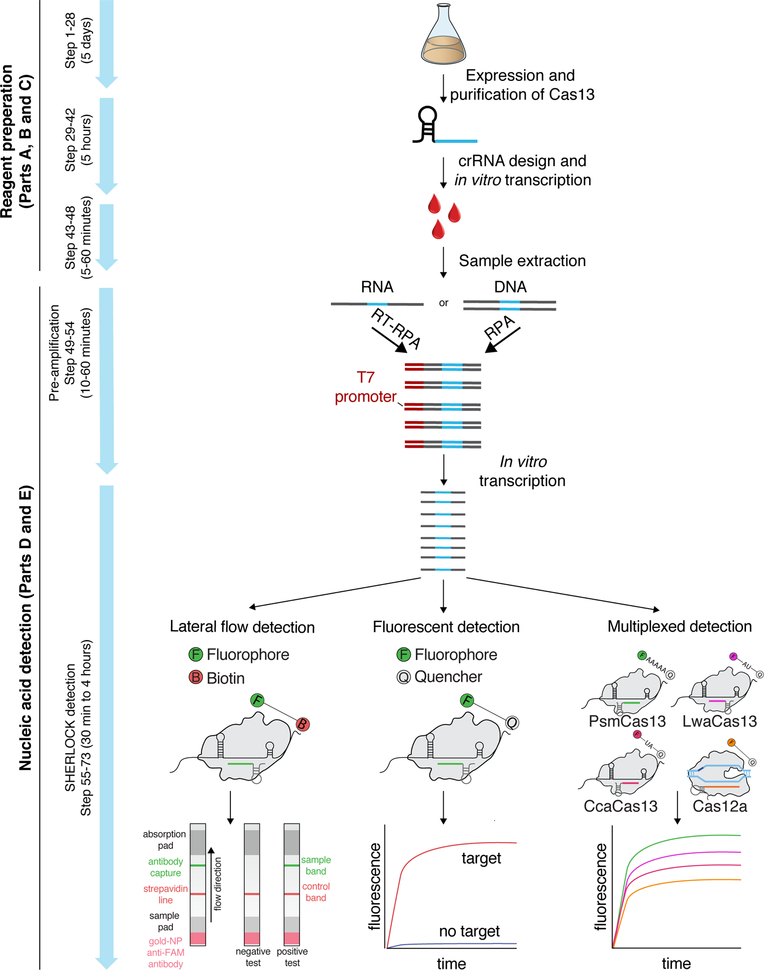
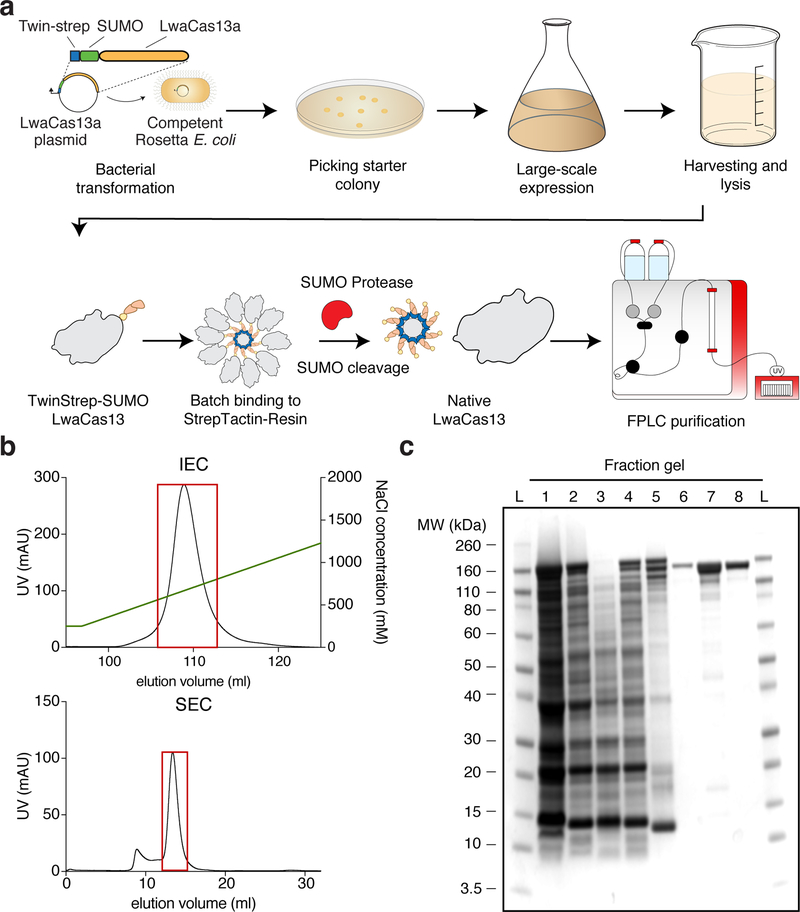
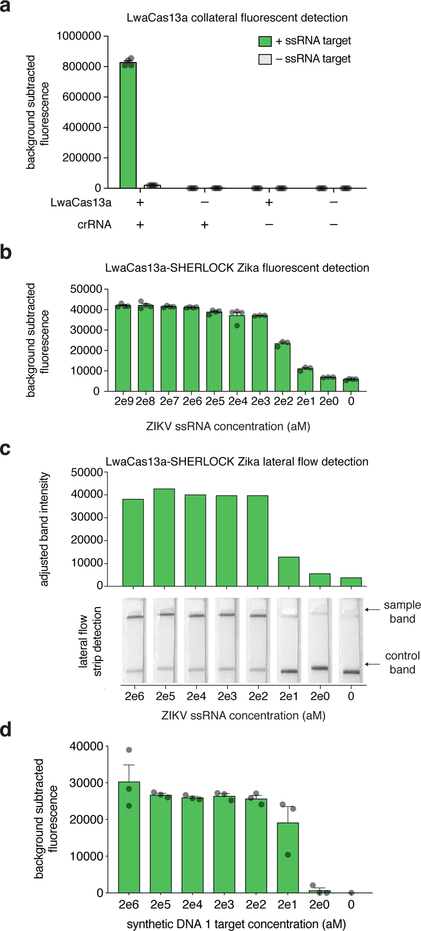
Rapid detection of nucleic acids is integral to applications in clinical diagnostics and biotechnology. We have recently established a CRISPR-based diagnostic platform that combines nucleic acid pre-amplification with CRISPR-Cas enzymology for specific recognition of desired DNA or RNA sequences. This platform, termed specific high-sensitivity enzymatic reporter unlocking (SHERLOCK), allows multiplexed, portable, and ultra-sensitive detection of RNA or DNA from clinically relevant samples. Here, we provide step-by-step instructions for setting up SHERLOCK assays with recombinase-mediated polymerase pre-amplification of DNA or RNA and subsequent Cas13- or Cas12-mediated detection via fluorescence and colorimetric readouts that provide results in <1 h with a setup time of less than 15 min. We also include guidelines for designing efficient CRISPR RNA (crRNA) and isothermal amplification primers, as well as discuss important considerations for multiplex and quantitative SHERLOCK detection assays.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
