

A framework for the investigation of rare genetic disorders in neuropsychiatry
- PMID: 31548702
- PMCID: PMC8656349
- DOI: 10.1038/s41591-019-0581-5
A framework for the investigation of rare genetic disorders in neuropsychiatry
Abstract
De novo and inherited rare genetic disorders (RGDs) are a major cause of human morbidity, frequently involving neuropsychiatric symptoms. Recent advances in genomic technologies and data sharing have revolutionized the identification and diagnosis of RGDs, presenting an opportunity to elucidate the mechanisms underlying neuropsychiatric disorders by investigating the pathophysiology of high-penetrance genetic risk factors. Here we seek out the best path forward for achieving these goals. We think future research will require consistent approaches across multiple RGDs and developmental stages, involving both the characterization of shared neuropsychiatric dimensions in humans and the identification of neurobiological commonalities in model systems. A coordinated and concerted effort across patients, families, researchers, clinicians and institutions, including rapid and broad sharing of data, is now needed to translate these discoveries into urgently needed therapies.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures


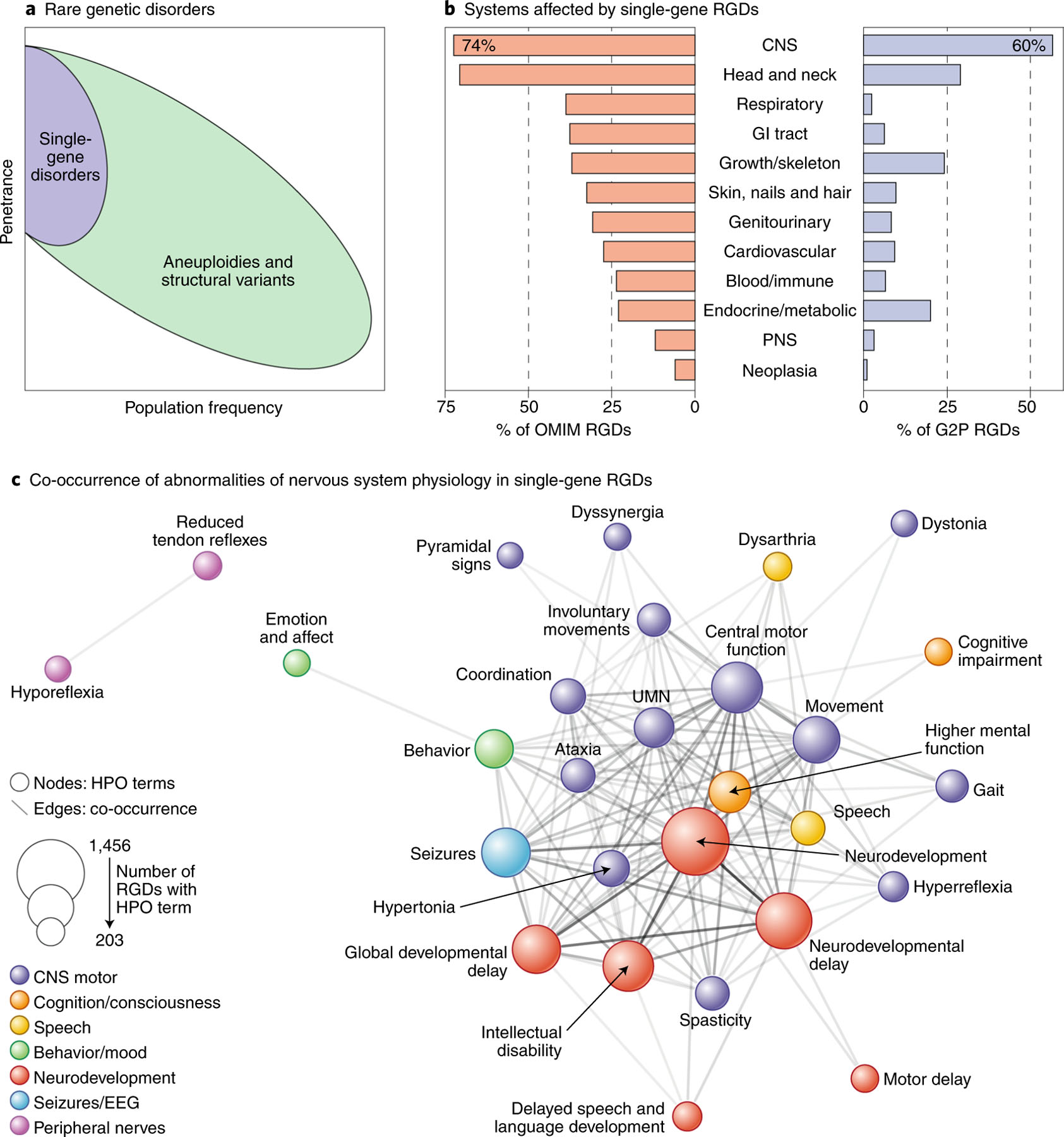
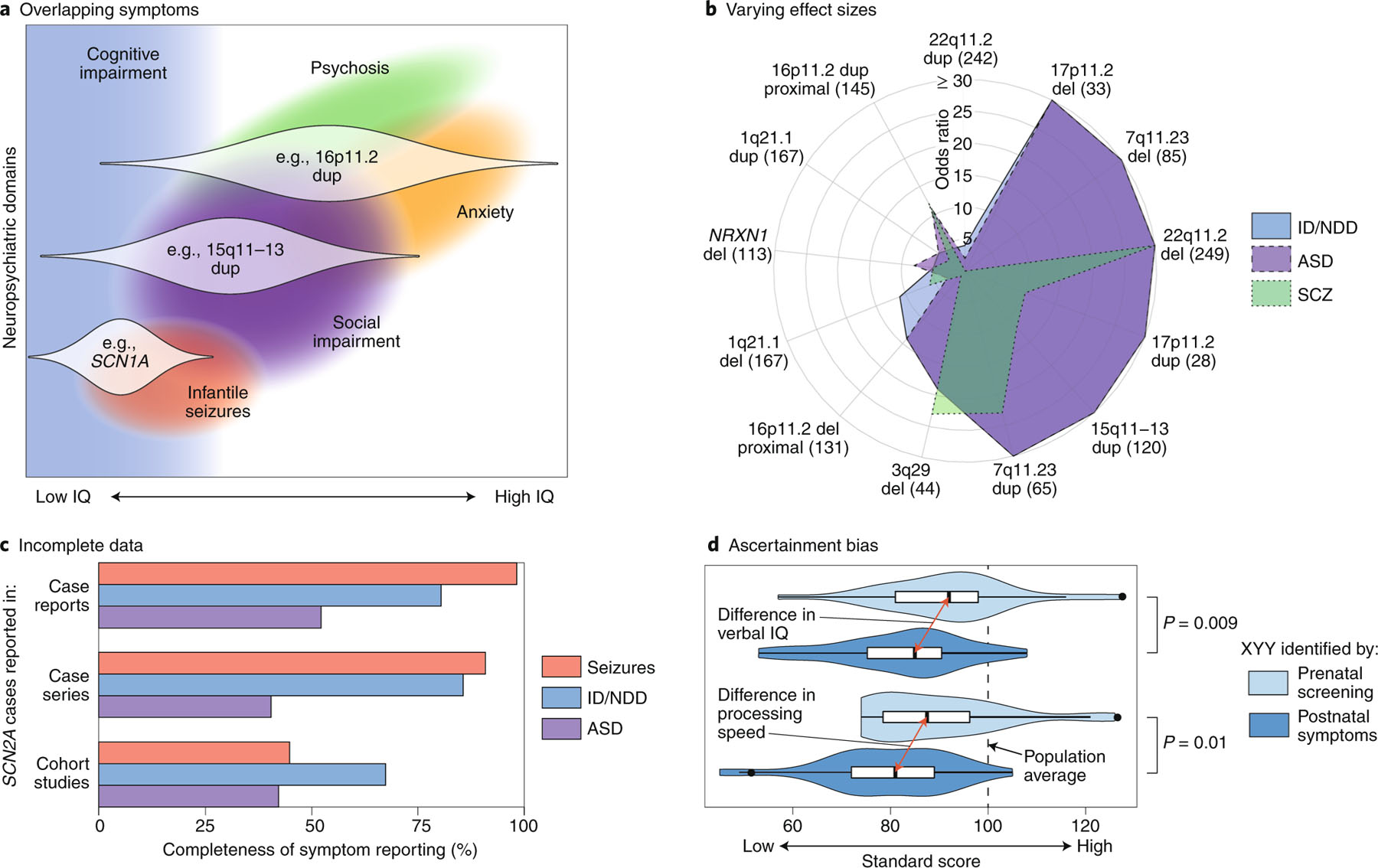
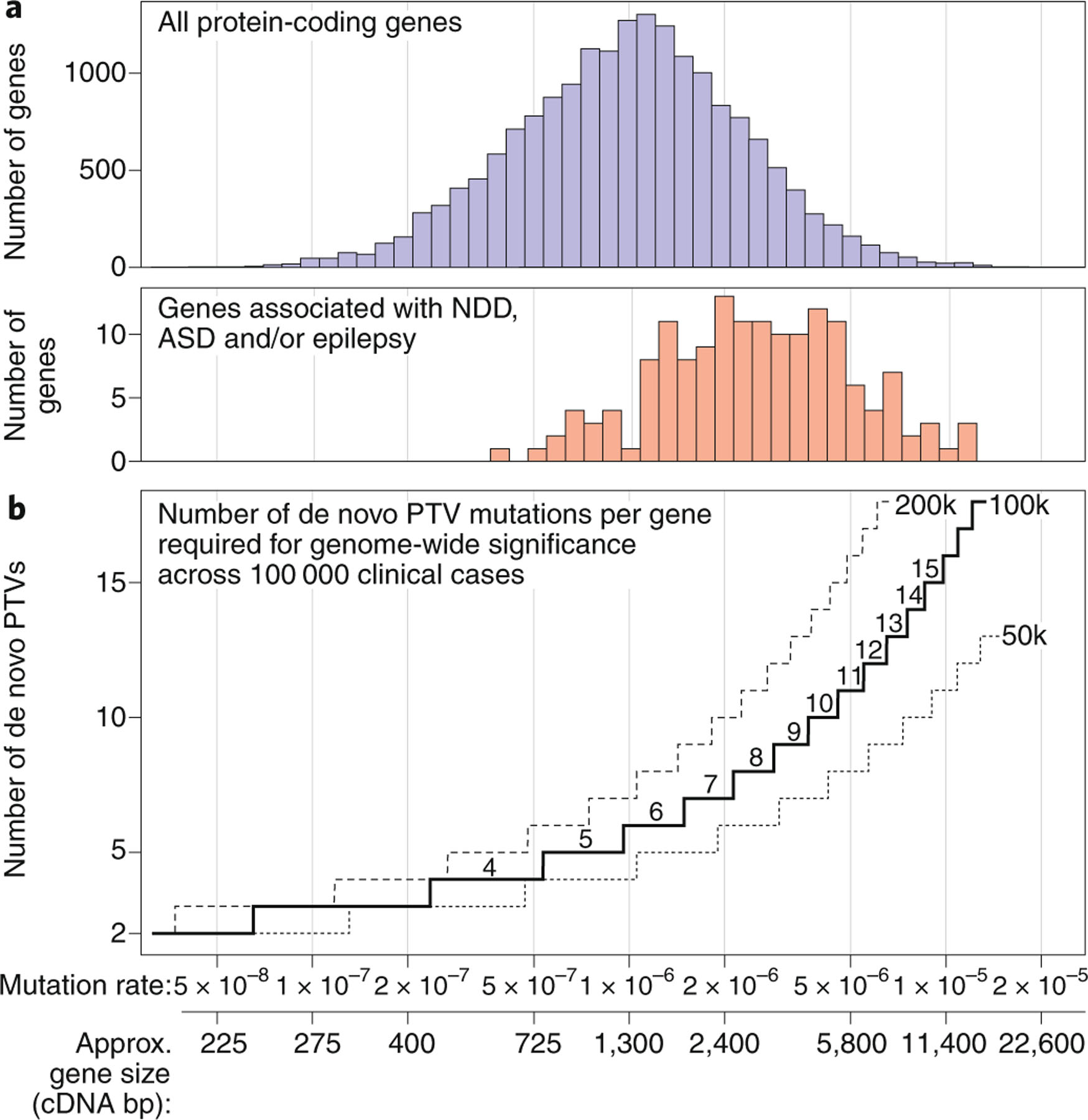
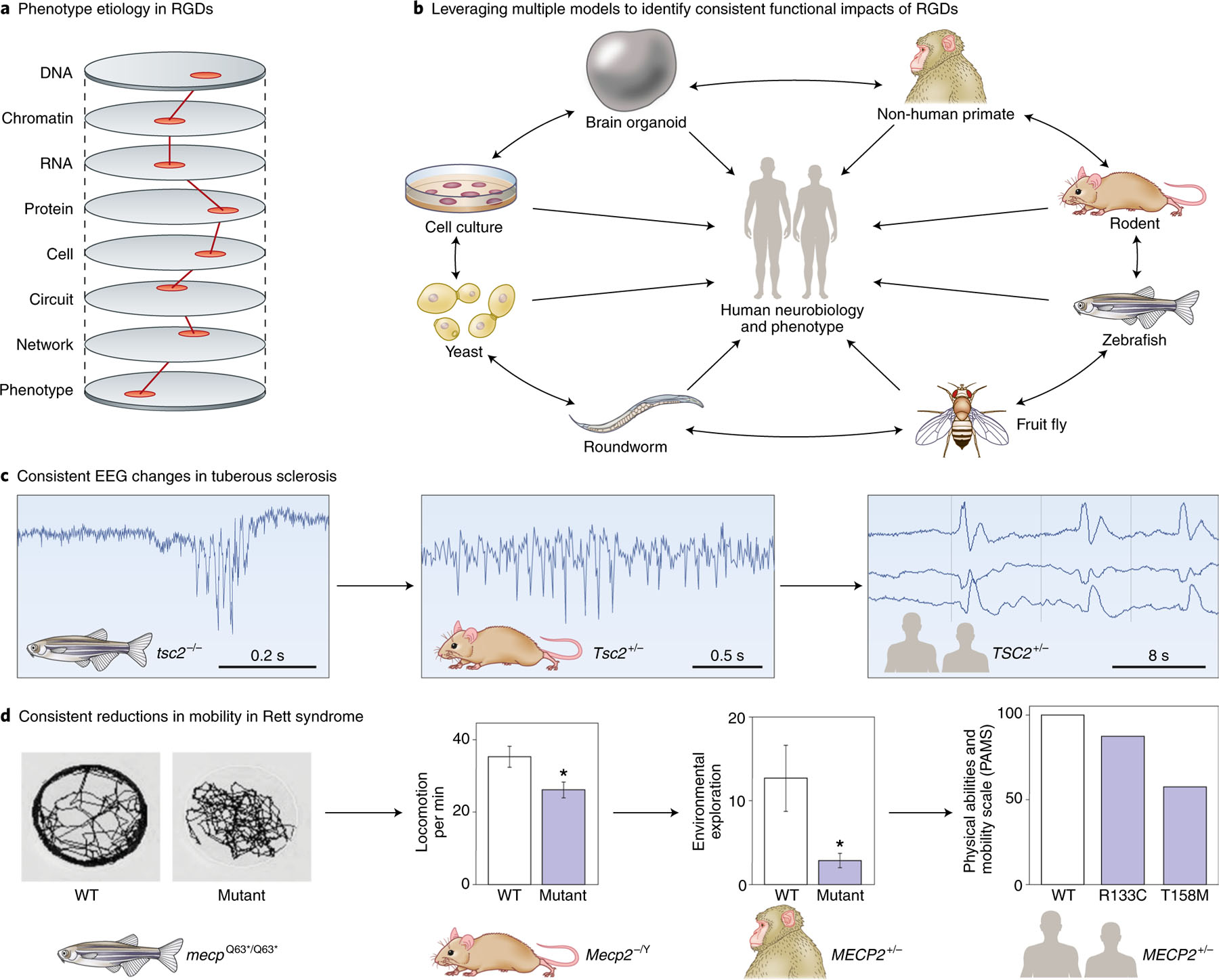
References
-
- US Food and Drug Administration. Orphan Drug Act. (1983).
-
- McKusick-Nathans Institute of Genetic Medicine. Online Mendelian Inheritance in Man, OMIM® (Johns Hopkins University, Baltimore, MD, USA: ) https://omim.org/ (accessed 28 April 2018).
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
