

Synergistic effects of common schizophrenia risk variants
- PMID: 31548722
- PMCID: PMC6778520
- DOI: 10.1038/s41588-019-0497-5
Synergistic effects of common schizophrenia risk variants
Abstract
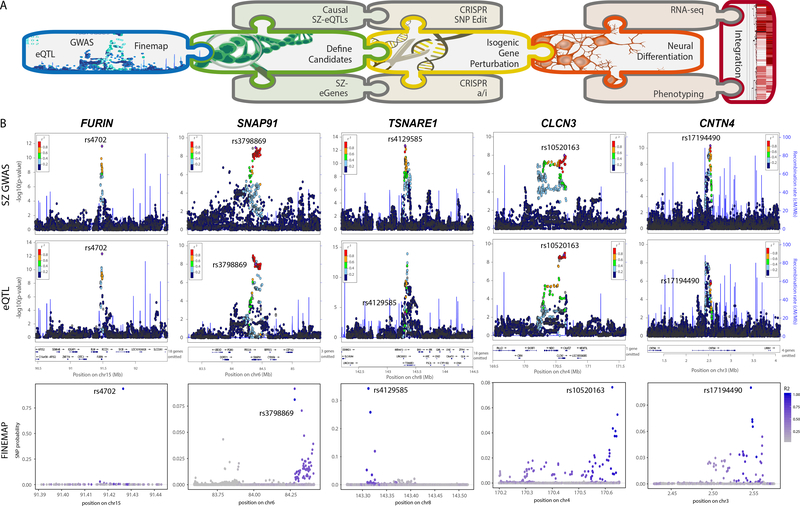
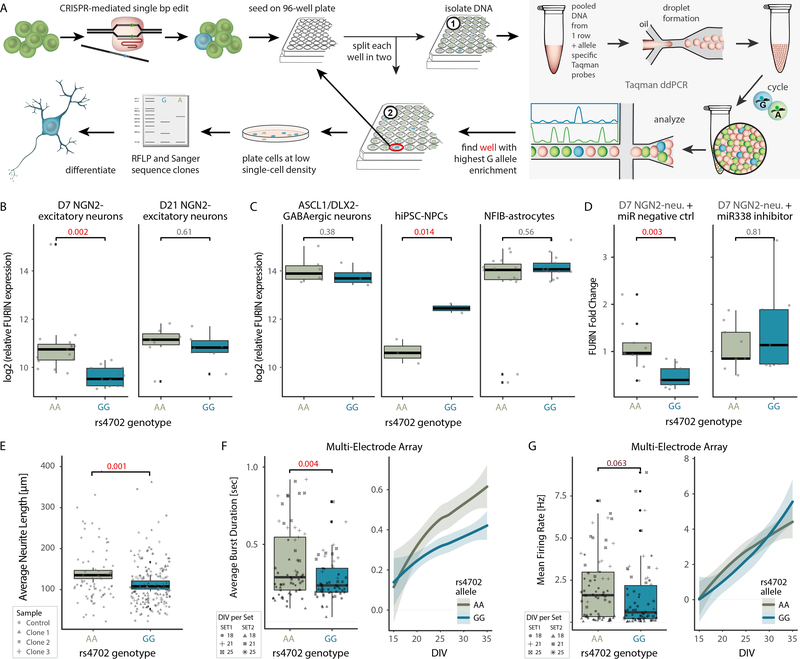
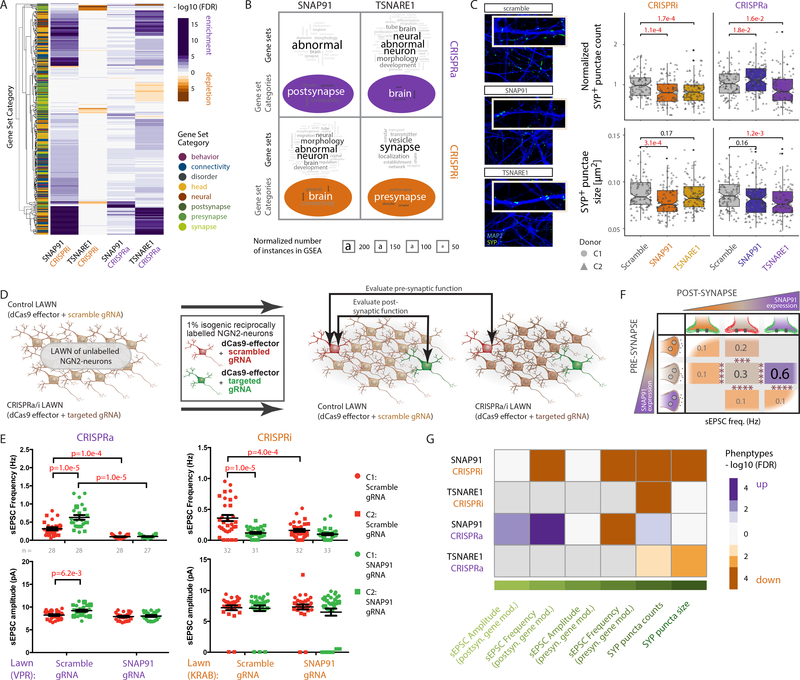
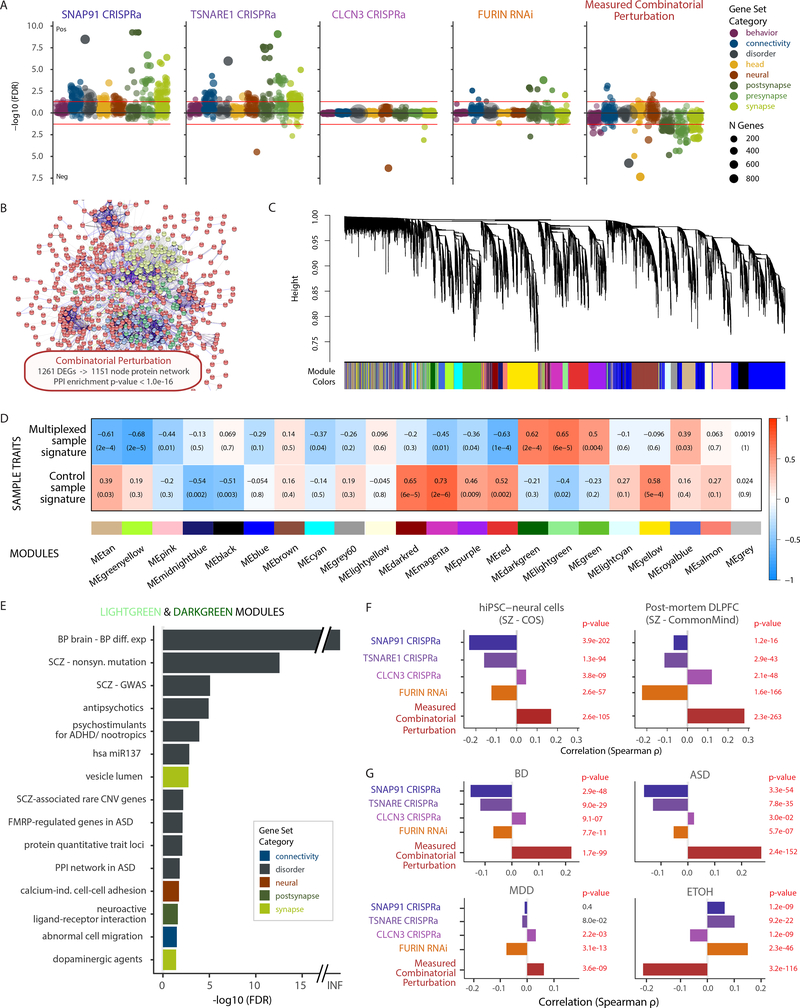
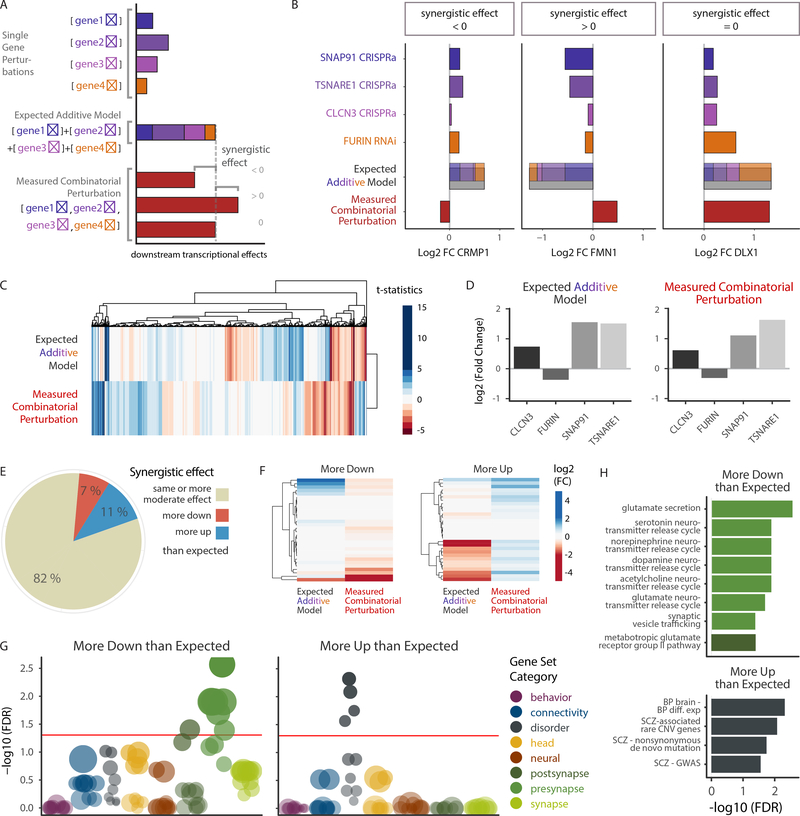
The mechanisms by which common risk variants of small effect interact to contribute to complex genetic disorders are unclear. Here, we apply a genetic approach, using isogenic human induced pluripotent stem cells, to evaluate the effects of schizophrenia (SZ)-associated common variants predicted to function as SZ expression quantitative trait loci (eQTLs). By integrating CRISPR-mediated gene editing, activation and repression technologies to study one putative SZ eQTL (FURIN rs4702) and four top-ranked SZ eQTL genes (FURIN, SNAP91, TSNARE1 and CLCN3), our platform resolves pre- and postsynaptic neuronal deficits, recapitulates genotype-dependent gene expression differences and identifies convergence downstream of SZ eQTL gene perturbations. Our observations highlight the cell-type-specific effects of common variants and demonstrate a synergistic effect between SZ eQTL genes that converges on synaptic function. We propose that the links between rare and common variants implicated in psychiatric disease risk constitute a potentially generalizable phenomenon occurring more widely in complex genetic disorders.
Conflict of interest statement
COMPETING FINANCIAL INTEREST STATEMENT
The authors declare no conflicts of interest.
Figures
Comment in
-
Modeling the cooperativity of schizophrenia risk genes.Nat Genet. 2019 Oct;51(10):1434-1436. doi: 10.1038/s41588-019-0508-6. Nat Genet. 2019. PMID: 31548721 No abstract available.
Similar articles
-
Schizophrenia-associated rs4702 G allele-specific downregulation of FURIN expression by miR-338-3p reduces BDNF production.Schizophr Res. 2018 Sep;199:176-180. doi: 10.1016/j.schres.2018.02.040. Epub 2018 Feb 28. Schizophr Res. 2018. PMID: 29499969
-
Gene expression elucidates functional impact of polygenic risk for schizophrenia.Nat Neurosci. 2016 Nov;19(11):1442-1453. doi: 10.1038/nn.4399. Epub 2016 Sep 26. Nat Neurosci. 2016. PMID: 27668389 Free PMC article.
-
A functional schizophrenia-associated genetic variant near the TSNARE1 and ADGRB1 genes.HGG Adv. 2024 Jul 18;5(3):100303. doi: 10.1016/j.xhgg.2024.100303. Epub 2024 May 3. HGG Adv. 2024. PMID: 38702885 Free PMC article.
-
An integrative functional genomics approach for discovering biomarkers in schizophrenia.Brief Funct Genomics. 2011 Nov;10(6):387-99. doi: 10.1093/bfgp/elr036. Epub 2011 Dec 8. Brief Funct Genomics. 2011. PMID: 22155586 Free PMC article. Review.
-
Path from schizophrenia genomics to biology: gene regulation and perturbation in neurons derived from induced pluripotent stem cells and genome editing.Neurosci Bull. 2015 Feb;31(1):113-27. doi: 10.1007/s12264-014-1488-2. Epub 2015 Jan 9. Neurosci Bull. 2015. PMID: 25575480 Free PMC article. Review.
Cited by
-
Schizophrenia risk alleles often affect the expression of many genes and each gene may have a different effect on the risk: A mediation analysis.Am J Med Genet B Neuropsychiatr Genet. 2021 Jun;186(4):251-258. doi: 10.1002/ajmg.b.32841. Epub 2021 Mar 8. Am J Med Genet B Neuropsychiatr Genet. 2021. PMID: 33683021 Free PMC article.
-
Eye-brain connections revealed by multimodal retinal and brain imaging genetics.Nat Commun. 2024 Jul 18;15(1):6064. doi: 10.1038/s41467-024-50309-w. Nat Commun. 2024. PMID: 39025851 Free PMC article.
-
Chromatin profiling in human neurons reveals aberrant roles for histone acetylation and BET family proteins in schizophrenia.Nat Commun. 2022 Apr 22;13(1):2195. doi: 10.1038/s41467-022-29922-0. Nat Commun. 2022. PMID: 35459277 Free PMC article.
-
Peripheral Blood Leukocyte RNA-Seq Identifies a Set of Genes Related to Abnormal Psychomotor Behavior Characteristics in Patients with Schizophrenia.Med Sci Monit. 2020 Feb 10;26:e922426. doi: 10.12659/MSM.922426. Med Sci Monit. 2020. PMID: 32038049 Free PMC article.
-
The polygenic architecture of schizophrenia - rethinking pathogenesis and nosology.Nat Rev Neurol. 2020 Jul;16(7):366-379. doi: 10.1038/s41582-020-0364-0. Epub 2020 Jun 11. Nat Rev Neurol. 2020. PMID: 32528109 Review.
References
METHODS REFERENCES
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
