

Psychiatric and Cognitive Aspects of Phenylketonuria: The Limitations of Diet and Promise of New Treatments
- PMID: 31551819
- PMCID: PMC6748028
- DOI: 10.3389/fpsyt.2019.00561
Psychiatric and Cognitive Aspects of Phenylketonuria: The Limitations of Diet and Promise of New Treatments
Abstract
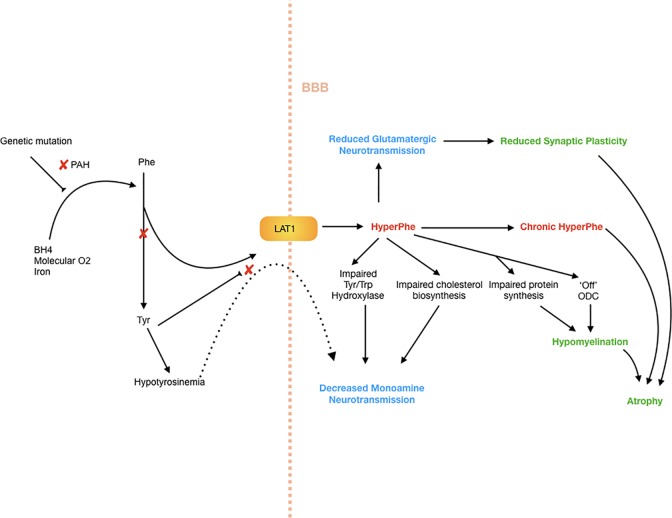
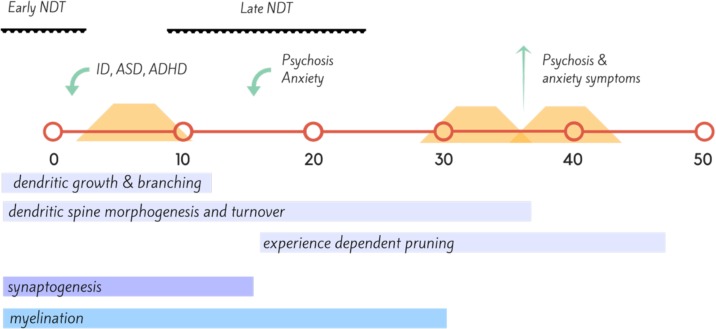
Phenylketonuria (PKU) is a recessive disorder of phenylalanine metabolism due to mutations in the gene for phenylalanine hydroxylase (PAH). Reduced PAH activity results in significant hyperphenylalaninemia, which leads to alterations in cerebral myelin and protein synthesis, as well as reduced levels of serotonin, dopamine, and noradrenaline in the brain. When untreated, brain development is grossly disrupted and significant intellectual impairment and behavioral disturbance occur. The advent of neonatal heel prick screening has allowed for diagnosis at birth, and the institution of a phenylalanine restricted diet. Dietary treatment, particularly when maintained across neurodevelopment and well into adulthood, has resulted in markedly improved outcomes at a cognitive and psychiatric level for individuals with PKU. However, few individuals can maintain full dietary control lifelong, and even with good control, an elevated risk remains of-in particular-mood, anxiety, and attentional disorders across the lifespan. Increasingly, dietary recommendations focus on maintaining continuous dietary treatment lifelong to optimize psychiatric and cognitive outcomes, although the effect of long-term protein restricted diets on brain function remains unknown. While psychiatric illness is very common in adult PKU populations, very little data exist to guide clinicians on optimal treatment. The advent of new treatments that do not require restrictive dietary management, such as the enzyme therapy Pegvaliase, holds the promise of allowing patients a relatively normal diet alongside optimized mental health and cognitive functioning.
Keywords: anxiety; cognitive function; depression; phenylketonuria; psychiatric.
Figures
References
-
- Scriver C, Kaufman S. Hyperphenylalaninemia: phenylalanine hydroxylase deficiency. In: the metabolic and molecular bases of inherited disease. McGraw-Hill Inc; (2001). p. 1667–724.
-
- Følling A. Uber Ausscheidung von Phenylbrenztraubensaure in den Harn als Stoffwechselanomalie in Vebindung mit Imbezzillitat. Hoppe-Seyler’s Z Physiol Chem (1934) 227:169–76. 10.1515/bchm2.1934.227.1-4.169 - DOI
-
- Jervis GA. Phenylpyruvic oligophrenia deficiency of phenylalanine-oxidizing system. Proc Soc Exp Biol Med (1953) 82(3):514–5. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases