

A Metataxonomic Tool to Investigate the Diversity of Treponema
- PMID: 31552004
- PMCID: PMC6746968
- DOI: 10.3389/fmicb.2019.02094
A Metataxonomic Tool to Investigate the Diversity of Treponema
Erratum in
-
Corrigendum: A Metataxonomic Tool to Investigate the Diversity of Treponema.Front Microbiol. 2019 Nov 8;10:2581. doi: 10.3389/fmicb.2019.02581. eCollection 2019. Front Microbiol. 2019. PMID: 31762772 Free PMC article.
Abstract
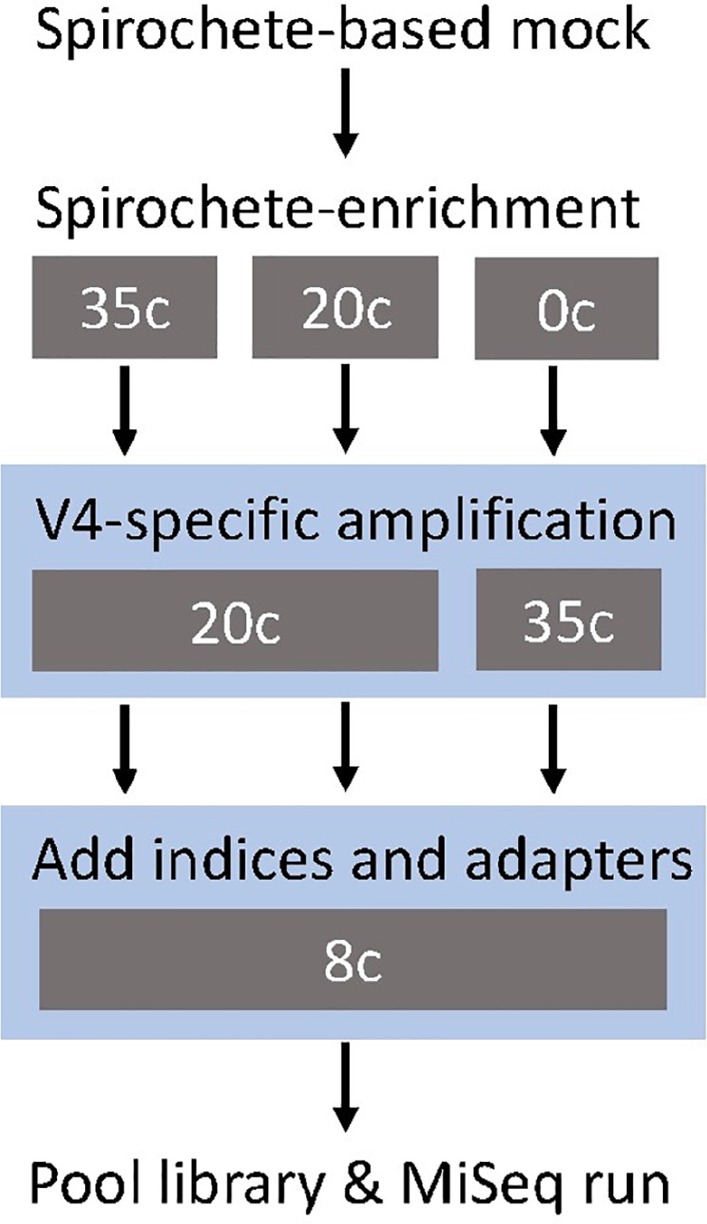
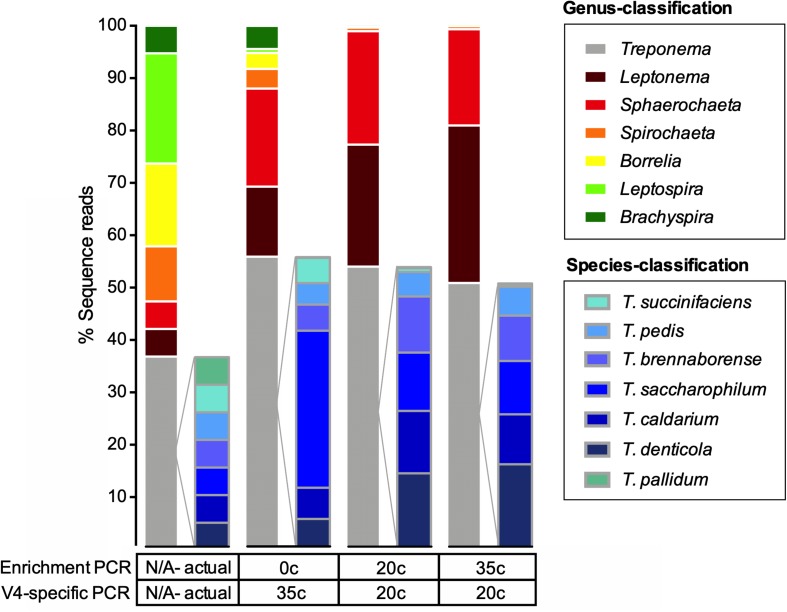
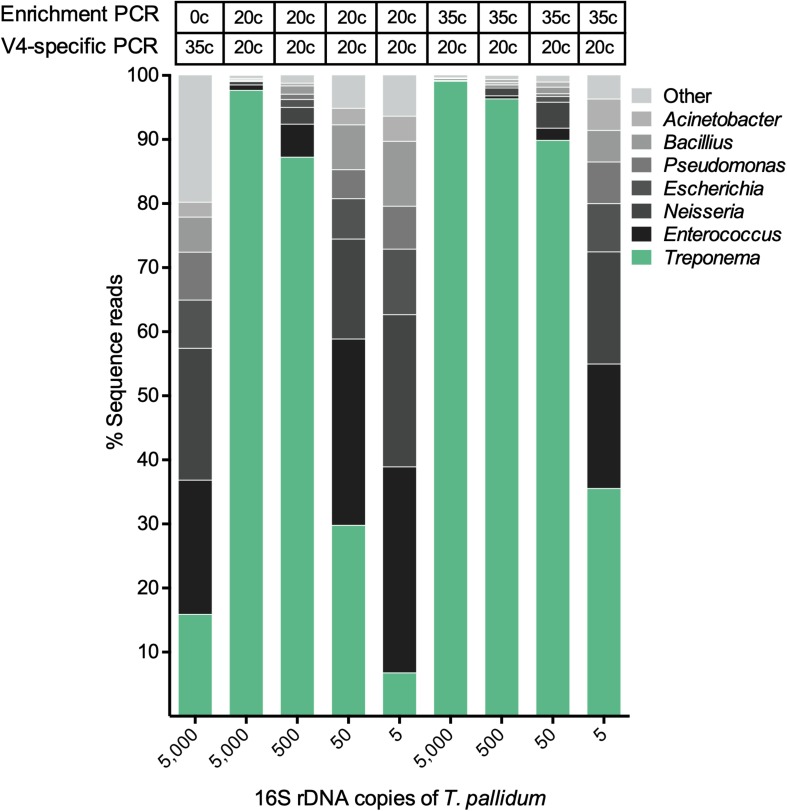
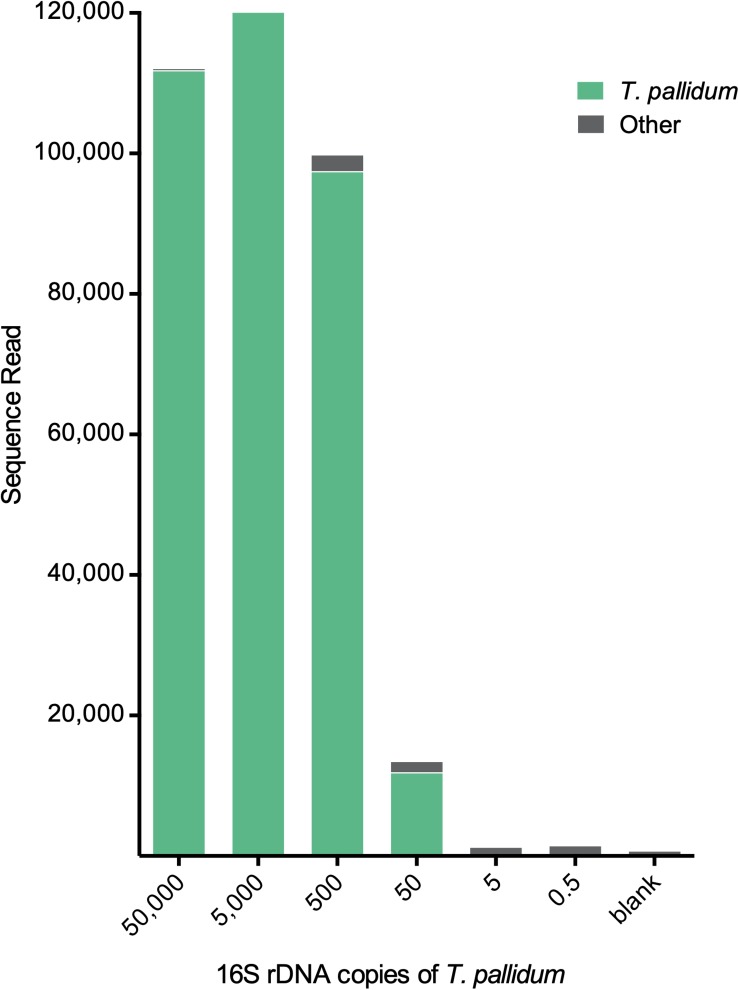
The genus Treponema contains a number of human and animal pathogenic as well as symbiotic bacteria that are found in vastly different anatomical and environmental habitats. Our understanding of the species range, evolution, and biology of these important bacteria is still limited. To explore the diversity of treponemes, we established, validated, and tested a novel metataxonomic approach. As the informative nature of the hypervariable regions of the 16S rRNA gene differ, we first analyzed each variable region independently. Considering the in silico results obtained, we established and validated the sequencing of the V4-region of the 16S rRNA gene using known mixtures of Treponema species as well as a selected number of clinical samples. The metataxonomic approach was able to identify Treponema to a near-species level. We demonstrate that using a spirochete-specific enrichment, our method is applicable to complex microbial communities and large variety of biological samples. The metataxonomic approach described provides a useful method to unravel the full diversity and range of Treponema in various ecosystems.
Keywords: 16S rRNA; Potorous; Treponema; marsupial; metagenomics; metataxonomics; one health; spirochete.
Figures
References
LinkOut - more resources
Full Text Sources
Research Materials