

Cross-Species Analysis of Single-Cell Transcriptomic Data
- PMID: 31552245
- PMCID: PMC6743501
- DOI: 10.3389/fcell.2019.00175
Cross-Species Analysis of Single-Cell Transcriptomic Data
Abstract
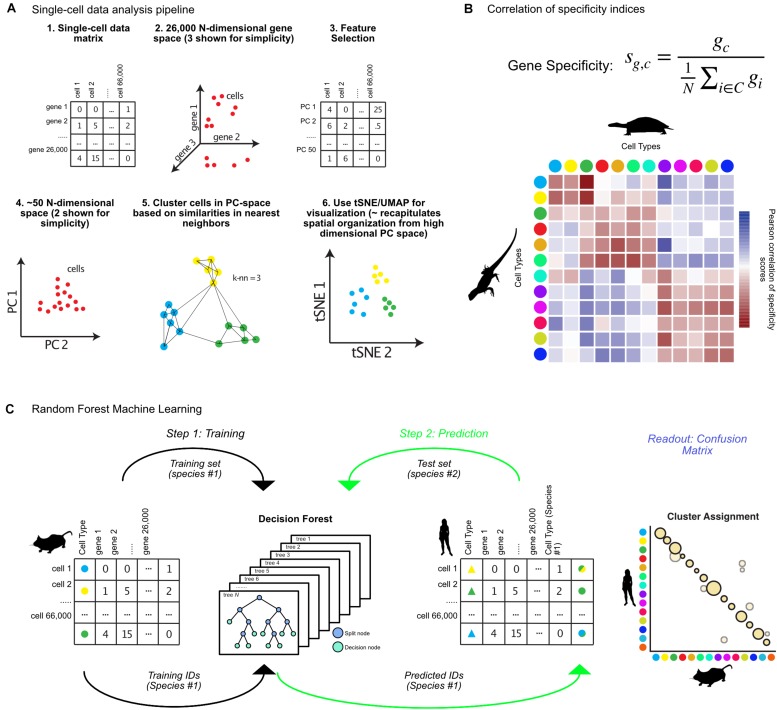
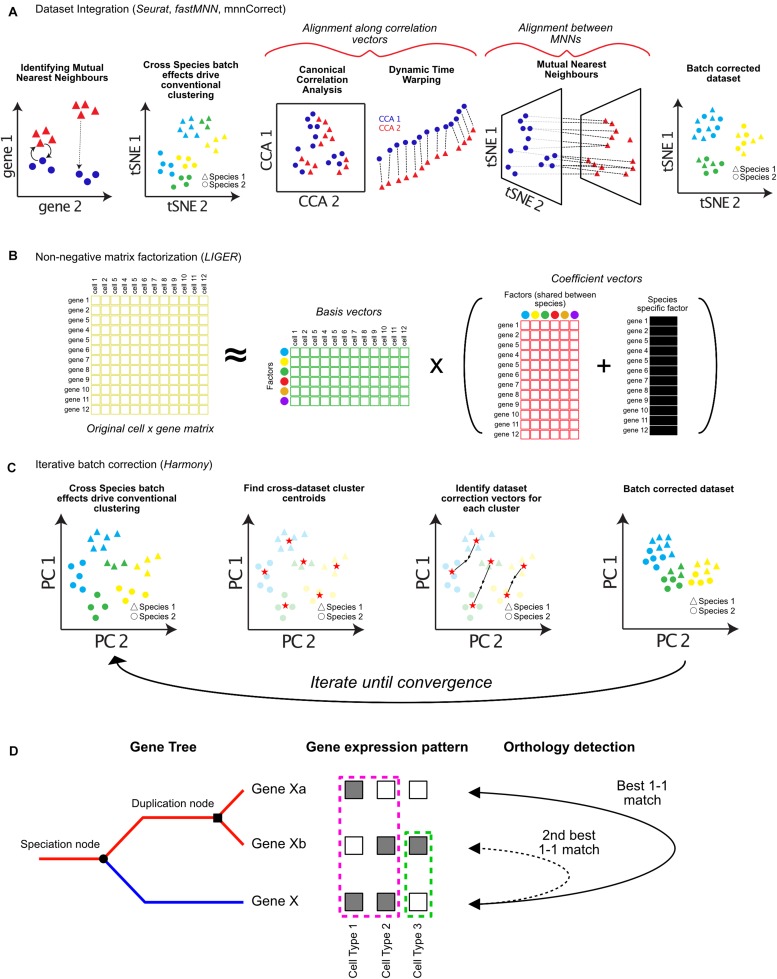
The ability to profile hundreds of thousands to millions of single cells using scRNA-sequencing has revolutionized the fields of cell and developmental biology, providing incredible insights into the diversity of forms and functions of cell types across many species. These technologies hold the promise of developing detailed cell type phylogenies which can describe the evolutionary and developmental relationships between cell types across species. This will require sampling of many species and taxa using single-cell transcriptomics, and methods to classify cell type homologies and diversifications. Many tools currently exist for analyzing single cell data and identifying cell types. However, cross-species comparisons are complicated by many biological and technical factors. These factors include batch effects common to deep-sequencing approaches, well known evolutionary relationships between orthologous and paralogous genes, and less well-understood evolutionary forces shaping transcriptome variation between species. In this review, I discuss recent developments in computational methods for the comparison of single-cell-omic data across species. These approaches have the potential to provide invaluable insight into how evolutionary forces act at the level of the cell and will further our understanding of the evolutionary origins of animal and cellular diversity.
Keywords: cell types; evolutionary cell biology; single-cell RNA sequencing; species comparisons; transcriptome evolution.
Figures
References
LinkOut - more resources
Full Text Sources