

Exon size and sequence conservation improves identification of splice-altering nucleotides
- PMID: 31554659
- PMCID: PMC6859846
- DOI: 10.1261/rna.070987.119
Exon size and sequence conservation improves identification of splice-altering nucleotides
Abstract
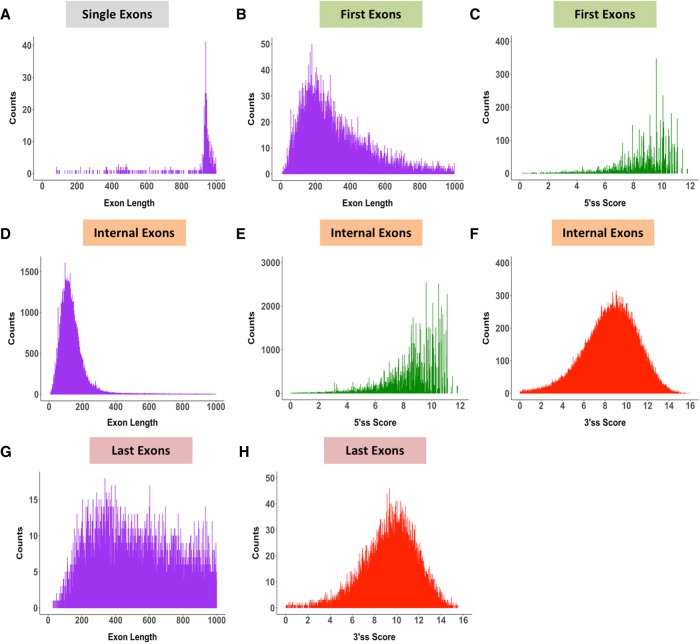
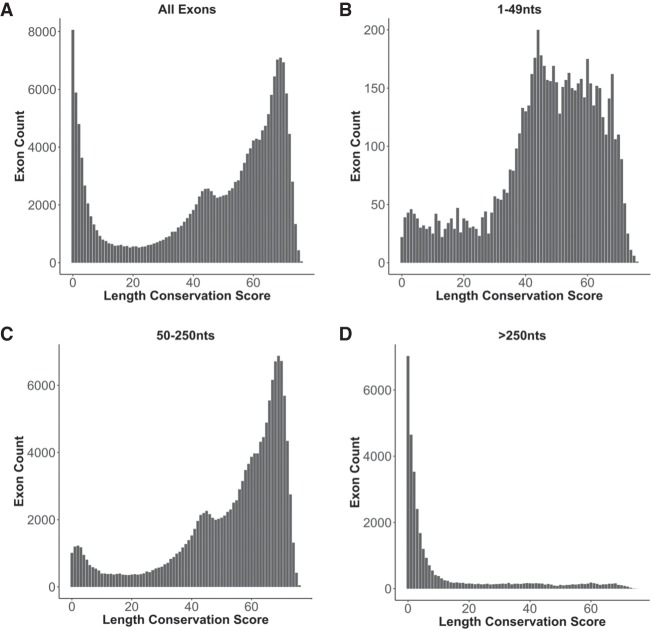
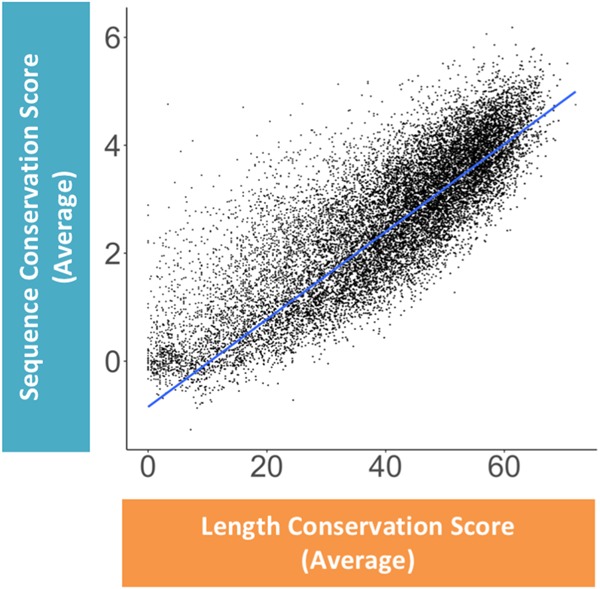
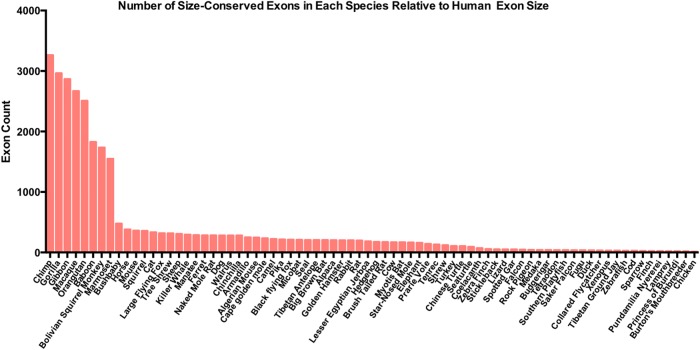
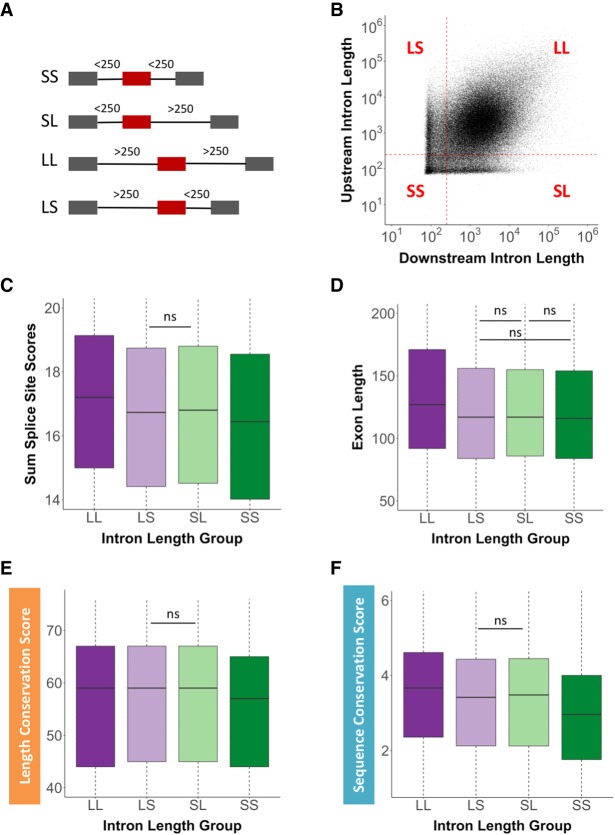
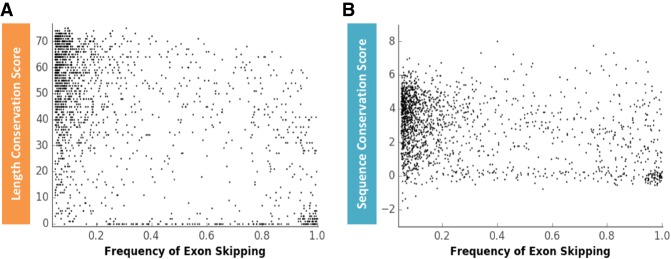
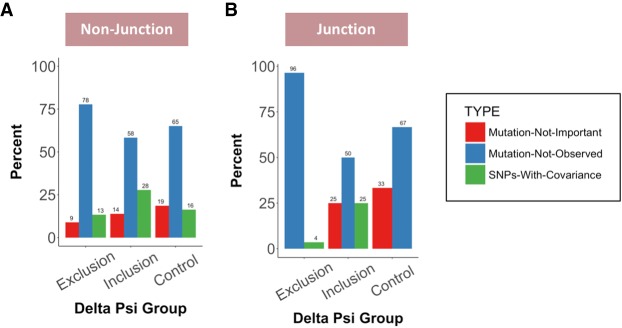
Pre-mRNA splicing is regulated through multiple trans-acting splicing factors. These regulators interact with the pre-mRNA at intronic and exonic positions. Given that most exons are protein coding, the evolution of exons must be modulated by a combination of selective coding and splicing pressures. It has previously been demonstrated that selective splicing pressures are more easily deconvoluted when phylogenetic comparisons are made for exons of identical size, suggesting that exon size-filtered sequence alignments may improve identification of nucleotides evolved to mediate efficient exon ligation. To test this hypothesis, an exon size database was created, filtering 76 vertebrate sequence alignments based on exon size conservation. In addition to other genomic parameters, such as splice-site strength, gene position, or flanking intron length, this database permits the identification of exons that are size- and/or sequence-conserved. Highly size-conserved exons are always sequence-conserved. However, sequence conservation does not necessitate exon size conservation. Our analysis identified evolutionarily young exons and demonstrated that length conservation is a strong predictor of alternative splicing. A published data set of approximately 5000 exonic SNPs associated with disease was analyzed to test the hypothesis that exon size-filtered sequence comparisons increase detection of splice-altering nucleotides. Improved splice predictions could be achieved when mutations occur at the third codon position, especially when a mutation decreases exon inclusion efficiency. The results demonstrate that coding pressures dominate nucleotide composition at invariable codon positions and that exon size-filtered sequence alignments permit identification of splice-altering nucleotides at wobble positions.
Keywords: SNPs; alternative splicing; exon conservation; phylogenetics; splicing.
© 2019 Movassat et al.; Published by Cold Spring Harbor Laboratory Press for the RNA Society.
Figures
References
-
- Berget SM. 1995. Exon recognition in vertebrate splicing. J Biol Chem 270: 2411–2414. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources